NECAT是肖传乐老师团队开发的一个针对Nanopore数据组装的软件,目前该工具尚未发表,除了https://github.com/xiaochuanle/NECAT 有软件的介绍外,暂时没有中文资料介绍NECAT的使用。
太长不看的结论 : Nanopore的组装推荐用下NECAT。组装之后是先用MEDAKA做一遍三代polish,然后用NextPolish默认参数做二代polish。
这篇将会以一篇发表在Nature Communication上的拟南芥nanopore数据介绍如何使用NECAT进行组装,运行在CentOS Linux release 7.3.1611 (Core),64G为内存, 20线程(Intel(R) Xeon(R) CPU E5-2640 v4 @ 2.40GHz),下面是正文。
软件安装 NECAT可以在https://github.com/xiaochuanle/NECAT/releases/ 页面获取最新的软件下载地址,这里下载的是0.01版本。
1 2 3 wget https://github.com/xiaochuanle/NECAT/releases/download/v0.01/necat_20190307_linux_amd64.tar.gz tar xzvf necat_20190307_linux_amd64.tar.gz export PATH=$PATH :$(pwd )/NECAT/Linux-amd64/bin
目前0.01版本不支持gz文件作为输入,但后续版本应该会支持。
目前更新到necat_20200119_Linux-amd64,新版本安装方法为
1 2 3 4 wget https://github.com/xiaochuanle/NECAT/releases/download/SourceCodes20200119/necat_20200119_Linux-amd64.tar.gz tar xzvf necat_20200119_Linux-amd64.tar.gz cd NECAT/Linux-amd64/bin$ export PATH=$PATH :$(pwd )
新版本增加gz文件支持。目前测试发现文件名需要符合xxx.fastq或xxx.fastq.gz命名格式,对于fq.gz无法识别,会导致程序文件出错。
实战 第一步 : 新建一个分析项目
以发表在NC上的拟南芥数据为例, 下载该数据
1 2 3 wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR217/003/ERR2173373/ERR2173373.fastq.gz seqkit seqkit fq2fa ERR2173373.fastq.gz | gzip -c > ERR2173373.fasta
第二步 : 创建配置文件
1 necat.pl config ath_config.txt
配置文件中,主要修改如下几个参数
1 2 3 4 5 6 PROJECT=athaliana ONT_READ_LIST=read_list.txt GENOME_SIZE=120000000 THREADS=20 MIN_READ_LENGTH=3000 CNS_OUTPUT_COVERAGE=45
参数中还有一个,NUM_ITER=2,它并非是简单的重复2次纠错,它的每一轮的校正目的其实不同,第一轮的优先级是敏感度(senstitive), 第二轮之后主要追求速度(fast)。
除了上面的配置参数外,其他参数可以不需要修改,使用默认的值即可。需要修改的话,参考最后的参数说明部分。
第三步 : 序列纠错
1 necat.pl correct ath_config.txt &
纠错后的reads在athaliana/1-consensus/cns_final.fasta
cns_finla.fasta的统计信息会输出在屏幕中, 或者自己用fsa_rd_stat也能得到同样的结果
1 2 3 4 5 6 7 8 9 10 Count: 206342 Tatal: 3102480870 Max: 112992 Min: 1010 N25: 31940 L25: 18989 N50: 21879 L50: 48506 N75: 13444 L75: 93215
此外我还用time获取了运行时间,纠错花了大概一个小时。
1 2 3 real 55m31.451s user 815m32.801s sys 7m55.039s
第四步 : contig组装
1 necat.pl assemble ath_config.txt &
结果在athaliana/4-fsa/contigs.fasta
关于contigs.fata统计信息会输出在屏幕上,同样用fsa_rd_stat 也可以。
1 2 3 4 5 6 7 8 9 10 Count: 162 Tatal: 122293198 Max: 14562810 Min: 1214 N25: 13052494 L25: 3 N50: 9503368 L50: 5 N75: 4919866 L75: 10
时间用了75分钟
1 2 3 real 74m53.127s user 1308m29.534s sys 12m5.032s
第五步 : contig搭桥
1 necat.pl bridge ath_config.txt
结果在athaliana/6-bridge_contigs/bridged_contigs.fasta
1 2 3 4 5 6 7 8 9 10 Count: 127 Tatal: 121978724 Max: 14562810 Min: 2217 N25: 13193939 L25: 3 N50: 11146374 L50: 5 N75: 5690371 L75: 9
从N50和N75可以看出这一步会提高组装的连续性。
组装结果polish 对Nanopore组装结果进行polish的常用软件有下面3个
由于拟南芥的基因组比较小,我分别用了Medaka和racon对输出结果进行polish(因为没有原始信号数据,因此nanopolish用不了),代码如下
Medaka
1 2 3 4 5 NPROC=20 BASECALLS=ERR2173373.fasta DRAFT=athaliana/6-bridge_contigs/bridged_contigs.fasta OUTDIR=medaka_consensus medaka_consensus -i ${BASECALLS} -d ${DRAFT} -o ${OUTDIR} -t ${NPROC} -m r941_min_high
三轮Racon:
1 2 3 4 5 6 7 gzip -dc ERR2173373.fastq.gz > ERR2173373.fastq minimap2 -t 20 ${DRAFT} ERR2173373.fastq > round_1.paf racon -t 20 ERR2173373.fastq round_1.paf ${DRAFT} > racon_round1.fasta minimap2 -t 20 racon_round1.fasta ERR2173373.fastq > round_2.paf racon -t 20 ERR2173373.fastq round_2.paf racon_round1.fasta> racon_round2.fasta minimap2 -t 20 racon_round2.fasta ERR2173373.fastq > round_3.paf racon -t 20 ERR2173373.fastq round_3.paf racon_round2.fasta> racon_round3.fasta
在后续评估质量的时候,我发现单纯用三代polish的结果还不是很好,因此我用他们提供的二代测序,用NextPolish对NECAT的结果进行polish。
1 2 3 prefetch ERR2173372 fasterq-dump -O . ERR2173372
run.cfg内容如下, 其中sgs.fofn记录的就是解压后的ERR2173372_1.fastq和ERR2173372_2.fastq的路径
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 [General] job_type = local job_prefix = nextPolish task = 1212 rewrite = no rerun = 3 parallel_jobs = 8 multithread_jobs = 20 genome = input.fasta genome_size = auto workdir = ./nextpolish polish_options = -p {multithread_jobs} [sgs_option] sgs_fofn = ./sgs.fofn sgs_options = -max_depth 100 -bwa
我考虑了两种情况,一种是直接用二代polish,另一种是三代polish之后接二代polish。
结果评估 在计算时间 上,我之前用Canu跑了相同的数据,设置原始错误率0.5,纠错后错误率为0.144,用3个节点(每个节点12个线程),运行了3天时间,但是NECAT只需要3个小时左右就能完成相同的分析,这个速度差异实在是太明显了。
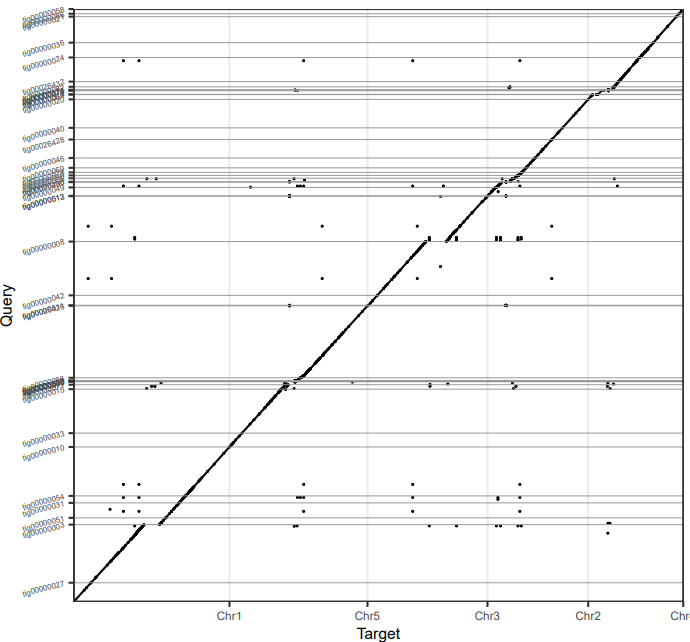
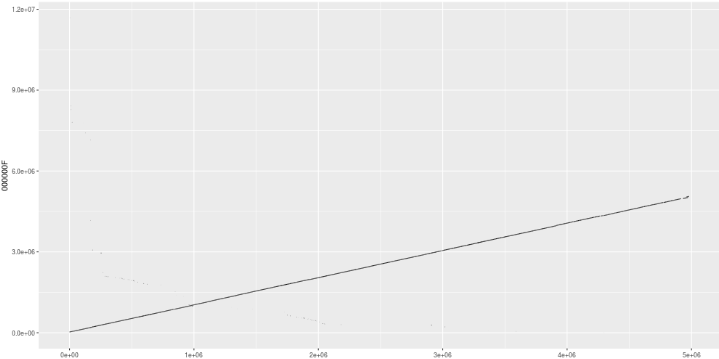
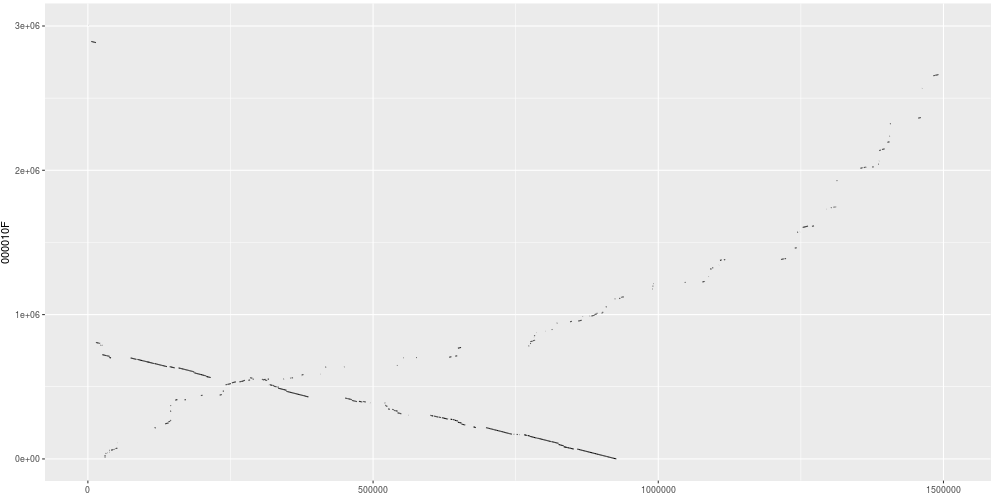
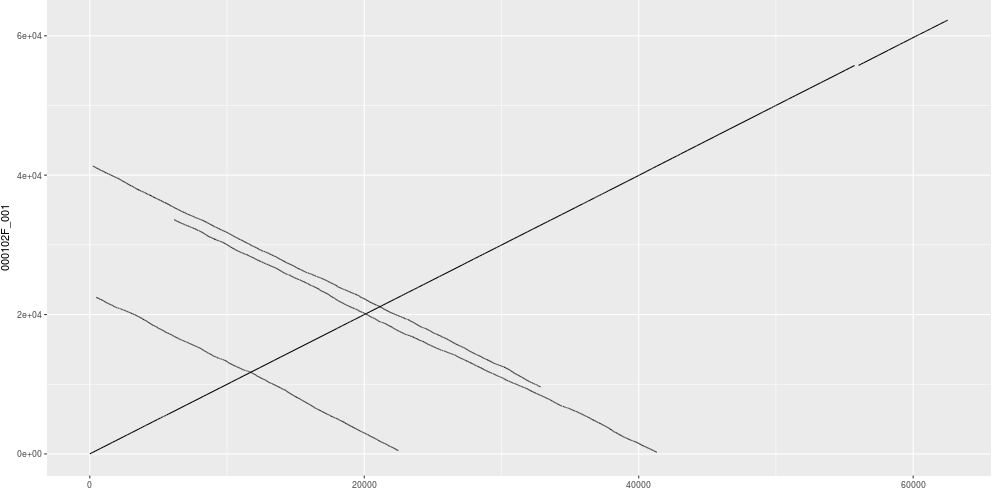
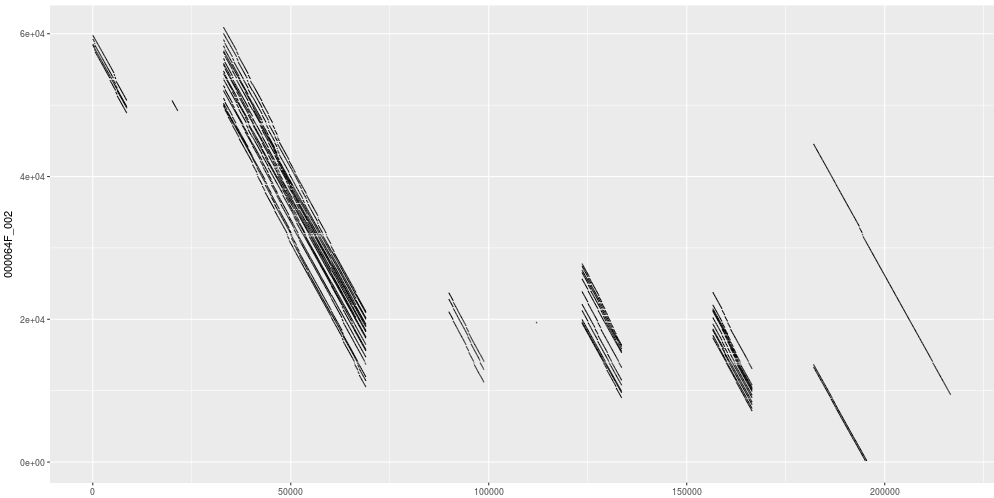
用Minimap2 + dotPlotly 绘制CANU,NECAT和拟南芥参考基因组的共线性图
1 2 3 4 minimap2 -t 20 -x asm5 Athaliana.fa NECAT.fa > NECAT.paf pafCoordsDotPlotly.R -i NECAT.paf -o NECAT -l -p 10 -k 5 minimap2 -t 20 -x asm5 Athaliana.fa CANU.fa > CANU.paf pafCoordsDotPlotly.R -i CANU.paf -o CANU -l -p 10 -k 5
NECAT的结果
CANU的结果
NECAT和CANU都和参考基因组有着良好的共线性,但是NECAT的连续性更好,几乎成一条直线。
之后,我使用了QUAST 来评估Canu,NECAT初步组装,NECAT用Medaka, nanopolish和racon纠错的结果(MD: MEDAKA, RC: RACON, NP:NextPolish)。
1 2 3 4 5 6 7 8 9 10 quast.py -t 100 --output-dir athaliana --circos \ CANU.fa \ NECAT.fa \ NECAT_MD.fa \ NECAT_MD_NP.fa \ NECAT_NP.fa \ NECAT_RC.fa \ NECAT_RC_NP.fa \ -r Athaliana.fa \ -g TAIR10_GFF3_genes.gff &
一些描述基本信息
1 2 3 4 5 6 7 CANU N50 = 4875070, L50 = 7, Total length = 114689024, GC % = 36.09 NECAT N50 = 11146374, L50 = 5, Total length = 121978724, GC % = 36.50 NECAT_MD N50 = 11216803, L50 = 5, Total length = 122101599, GC % = 36.54 NECAT_MD_NP N50 = 11405151, L50 = 5, Total length = 124142955, GC % = 36.30 NECAT_NP N50 = 11399084, L50 = 5, Total length = 124735066, GC % = 36.36 NECAT_RC N50 = 11212098, L50 = 5, Total length = 122519370, GC % = 36.4 NECAT_RC_NP N50 = 11406553, L50 = 5, Total length = 124618502, GC % = 36.34
在BUSCO完整度上, 以embryophyta_odb10作为物种数据库, 其中ONTmin_IT4是发表的文章里的结果, Athalina则是拟南芥的参考基因组,我们以它们的BUSCO值作为参照。
1 2 3 4 5 6 Athalina : C:98.6%[S:98.0%,D:0.6%],F:0.4%, M:1.0%, n:1375 ONTmin_IT4 : C:98.4%[S:97.7%,D:0.7%],F:0.7%, M:0.9%, n:1375 CANU : C:22.9%[S:22.8%,D:0.1%],F:20.2%,M:56.9%,n:1375 NECAT : C:36.6%[S:36.6%,D:0.0%],F:22.9%,M:40.5%,n:1375 NECAT_MEDAKA : C:53.6%[S:53.2%,D:0.4%],F:21.0%,M:25.4%,n:1375 NECAT_RACON : C:45.3%[S:45.2%,D:0.1%],F:23.1%,M:31.6%,n:1375
二代Polish后的BUSCO结果如下(MD: MEDAKA, RC: RACON, NP:NextPolish):
1 2 3 4 5 Athalina : C:98.6%[S:98.0%,D:0.6%],F:0.4%,M:1.0%,n:1375 ONTmin_IT4 : C:98.4%[S:97.7%,D:0.7%],F:0.7%,M:0.9%,n:1375 NECAT_NP : C:98.6%[S:97.9%,D:0.7%],F:0.4%,M:1.0%,n:1375 NECAT_MD_NP: C:98.7%[S:98.0%,D:0.7%],F:0.4%,M:0.9%,n:1375 NECAT_RC_NP: C:98.5%[S:97.8%,D:0.7%],F:0.4%,M:1.1%,n:1375
从以上这些数据,你可以得到以下几个洞见:
在Nanopore的组装上,NECAT效果优于Canu,无论是连续性还是N50上
MEDAKA三代polish效果好于RACON。在速度上,MEDAKA比三遍RACON都慢,并且MEDAKA会将一些可能的错误组装给打断
Nanopore的数据用NECAT组装后似乎用NextPolish进行polish后就行,但是由于物种比较小,可能不具有代表性。
结论 : Nanopore的组装建议用NECAT。组装之后是先用MEDAKA做一遍三代polish,然后用NextPolish默认参数做二代polish。
配置文件补充 这部分对配置文件做一点简单补充。
下面这些参数相对简单,不需要过多解释,按照自己需求修改
CLEANUP: 运行完是否清理临时文件,默认是0,表示不清理
USE_GRID: 是否使用多节点, 默认是false
GRID_NODE: 使用多少个节点,默认是0,当USE_GRID为true时,按照自己实际情况设置
以下的参数则是需要根据到具体的软件中去查看具体含义,需要和软件开发者讨论
OVLP_FAST_OPTIONS: 第二轮纠错时, 传给oc2pmov
OVLP_SENSITIVE_OPTIONS: 第一轮纠错时, 传给oc2pmov
CNS_FAST_OPTIONS: 第二轮纠错时,传给oc2cns
CNS_SENSITIVE_OPTIONS: 第一轮纠错时,传给oc2cns
TRIM_OVLP_OPTIONS: 传给oc2asmpm
ASM_OVLP_OPTIONS: 传给oc2asmpm
FSA_OL_FILTER_OPTIONS: 参数传给fsa_ol_filter
FSA_ASSEMBLE_OPTIONS: 参数传给fsa_assemble
FSA_CTG_BRIDGE_OPTIONS: 参数传给fsa_ctg_bridge
参考资料