Canu简介
Canu是Celera的继任者,能用于组装PacBio和Nanopore两家公司得到的测序结果。
Canu分为三个步骤,纠错,修整和组装,每一步都差不多是如下几个步骤:
- 加载read到read数据库,gkpStore
- 对k-mer进行技术,用于计算序列间的overlap
- 计算overlap
- 加载overlap到overlap数据库,OvlStore
- 根据read和overlap完成特定分析目标
- read纠错时会从overlap中挑选一致性序列替换原始的噪声read
- read修整时会使用overlap确定read哪些区域是高质量区域,哪些区域质量较低需要修整。最后保留单个最高质量的序列块
- 序列组装时根据一致的overlap对序列进行编排(layout), 最后得到contig。
这三步可以分开运行,既可以用Canu纠错后结果作为其他组装软件的输入,也可以将其他软件的纠错结果作为Canu的输入,因此下面分别运行这三步,并介绍重要的参数。
几个全局参数:genomeSize设置预估的基因组大小,这用于让Canu估计测序深度; maxThreads设置运行的最大线程数;rawErrorRate用来设置两个未纠错read之间最大期望差异碱基数;correctedErrorRate则是设置纠错后read之间最大期望差异碱基数,这个参数需要在 组装 时多次调整;minReadLength表示只使用大于阈值的序列,minOverlapLength表示Overlap的最小长度。提高minReadLength可以提高运行速度,增加minOverlapLength可以降低假阳性的overlap。
组装实战
数据下载
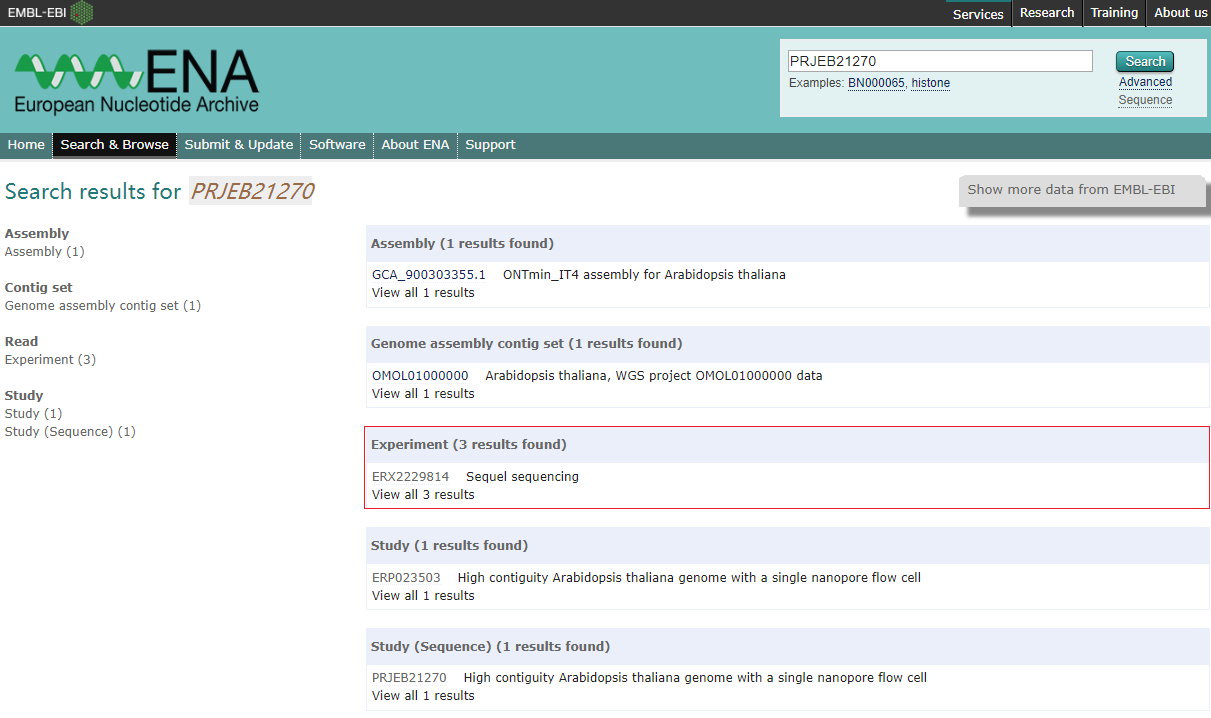
数据来自于发表在 Nature Communication 的一篇文章 “High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell“。 这篇文章提供了 Arabidopsis thaliana KBS-Mac-74 的30X短片段文库二代测序、PacBio和Nanopore的三代测序以及Bionano测序数据, 由于拟南芥的基因组被认为是植物中的金标准,因此文章提供的数据适合非常适合用于练习。根据文章提供的项目编号”PRJEB21270”, 在European Nucleotide Archive上找到下载地址。
1 | ## PacBio Sequal |
下载的PacBio数据以BAM格式存储,可以通过安装PacBio的smrtlink工具套装,使用其中的bam2fasta工具进行转换.
1 | # build index for convert |
其实samtools fasta也可以将bam转成fasta文件,并且不影响之后的组装。
PacBio的smrtlink工具套装大小为1.4G,不但下载速度慢,安装也要手动确认各种我不清楚的选项, 唯一好处就是工具很全。
运行Canu
第一步:纠错。三代测序本身错误率高,使得原始数据充满了噪音。这一步就是通过序列之间的相互比较纠错得到高可信度的碱基。主要调整两个参数
- corOutCoverage: 用于控制多少数据用于纠错。比如说拟南芥是120M基因组,100X测序后得到了12G数据,如果只打算使用最长的6G数据进行纠错,那么参数就要设置为50(120m x 50)。设置一个大于测序深度的数值,例如120,表示使用所有数据。
1 | canu -correct \ |
可以将上述命令保存到shell脚本中进行运行, nohup bash run_canu.sh 2> correct.log &.
注: 1.7版本里会没有默认没有安装gnuplot出错,因此gnuplotTested=true 可以跳过检查。
第二步:修整。这一步的目的是为了获取更高质量的序列,移除可疑区域(如残留的SMRTbell接头).
1 | canu -trim \ |
第三步: 组装。在前两步获得高质量的序列后,就可以正式进行组装. 这一步主要调整的就是纠错后的序列的错误率, correctedErrorRate,它会影响utgOvlErrorRate。这一步可以尝试多个参数,因为速度比较块。
1 | # error rate 0.035 |
最后输出文件下的ath.contigs.fasta就是结果文件。
一些宝贵的建议
Nanopore组装
对于Nanopore数据,使用Canu组装并不是一个非常好的选择,我曾经以一个600多M的物种100X数据进行组装,在120线程,花了整整一个多月的时间,尽管它的组装效果真的是很好。
- rawErrorRate: 从0.144 调整到 0.12 或者更低,速度会提高5到10倍
- readSamplingCoverage, readSamplingBias: 可以抽取部分数据进行纠错,而不是全部数据
- corOutCoverage=30: 默认的30X或者40X的组装效果其实不错
-fast: 对于1Gb以下的物种,可以加上该参数,会明显提高速度
高杂合物种组装
对于高杂合物种的组装,Canu建议是用 batOptions=-dg 3 -db 3 -dr 1 -ca 500 -cp 50参数尽量分出两套单倍型,然后对基因组去冗余。
batOptions表示传递后续的参数给组装软件bogart, -dg 3 -db3降低自动确定阈值时的错误率离差(deviation),从而更好的分开单倍型。-dr 1 -ca 500 -cp 50会影响错误组装的拆分,对于一个模棱两可的contig,如果至少另一条可选路径的overlap长度至少时500bp,或者说另一条可选路径时在长度上和当前最佳路径存在50%的差异,那么就将contig进行拆分。
关于杂合物种组装的讨论,参考https://github.com/marbl/canu/issues/201#issuecomment-233750764
购买SSD避免服务器IO瓶颈
如果你的服务器线程数很多,你在普通的机械硬盘上运行组装,而且你的系统还是CentOS,那么你需要调整一个参数,避免其中一步的IO严重影响服务器性能。
Canu通过两个策略进行并行,bucketizing (‘ovb’ 任务) 和 sorting (‘ovs’ 任务)。 bucketizing会从1-overlap读取输出的overlap,将他们复制一份作为中间文件。sorting一步将这些文件加载到内存中进行排序然后写出到硬盘上。 如果你的overlap输出特别多,那么该步骤将会瞬间挤爆的你的IO.
为了避免悲剧发生,请增加如下参数: ovsMemory=16G ovbConcurrency=15 ovsConcurrency=15, 也就是降低这两步同时投递的任务数,缓解IO压力。
如何用不同服务器处理同一个任务
overlap这一步时间久,如果并没有服务器集群,而是有多台服务器,可以参考如下方法进行数据并行处理(必须要安装相同的Canu版本)
假如Canu任务中的prefix参数为coc, 用scp进行数据的传递
1 | scp -r coc.seqStore wangjw@10.10.87.132:/data1/wjw/Coc/Canu |
到目标服务器的canu目录下
1 | mkdir unitigging |
修改overlap.sh里的bin路径,指定当前服务器的canu路径
之后运行任务即可
1 | cd unitigging/1-overlapper/ |
运行完任务之后,将目前服务器unitigging/1-overlapper/001目录下的000077.oc 000077.ovb 000077.stats文件传送回原来的服务器unitigging/1-overlapper/001下即可。