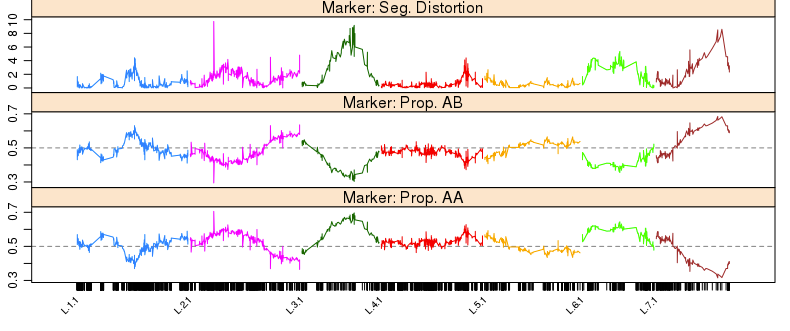
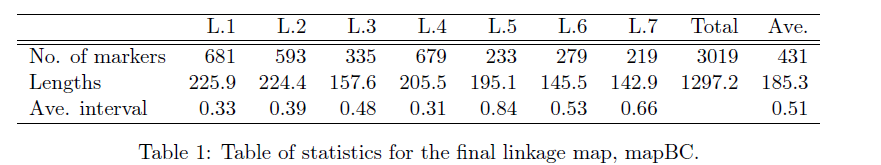
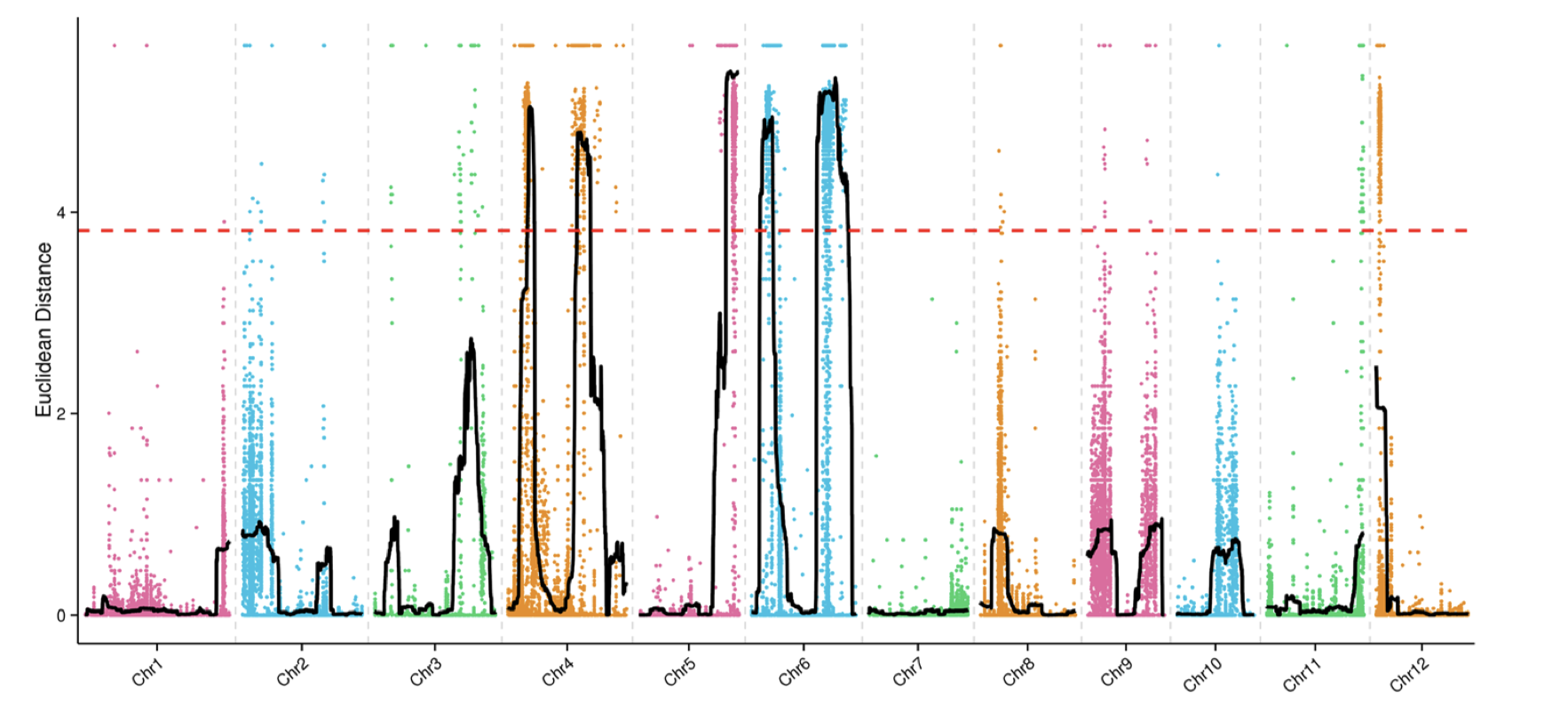
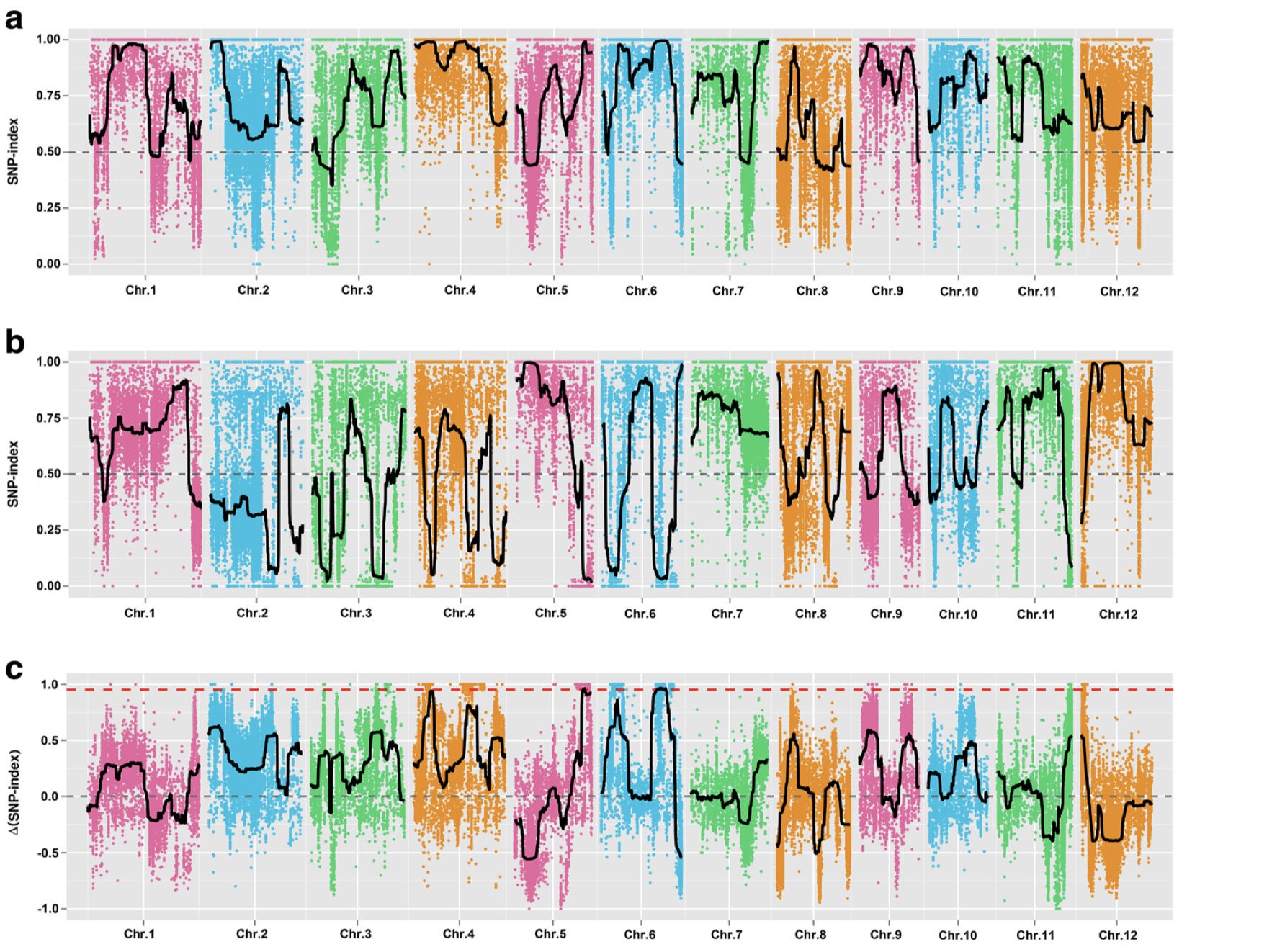
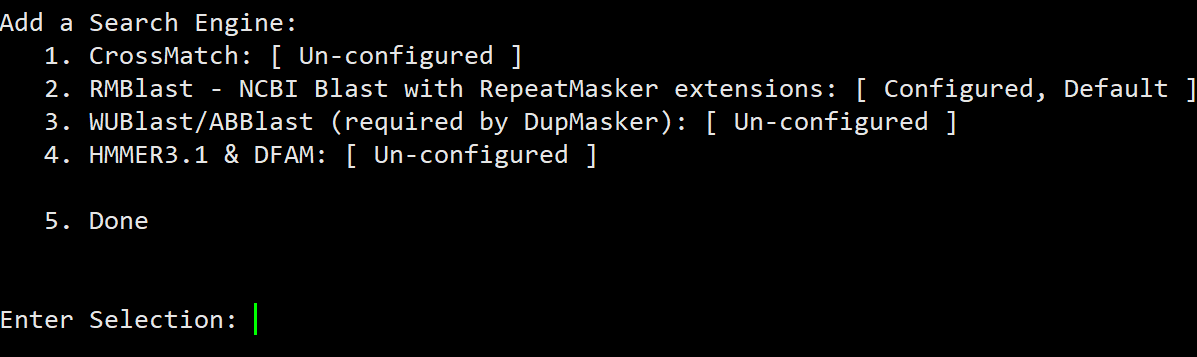
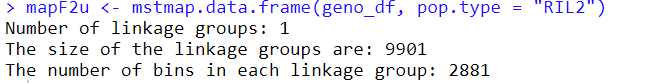在遗传定位上,相对于GWAS和binmap,BSA是一个比较省钱的策略,它只需要测两个亲本和后代中两个极端差异群体即可,但是它对实验设计,表型考察,样本挑选都有比较高的要求。如果你的表型差异并不是泾渭分明,那么还是不要用BSA比较合适。这方面知识,建议阅读徐云碧老师2016年发表在PBJ上的”Bulked sample analysis in genetics, genomics and crop improvement”, 这篇教程侧重于实际分析,而非理论讲解。
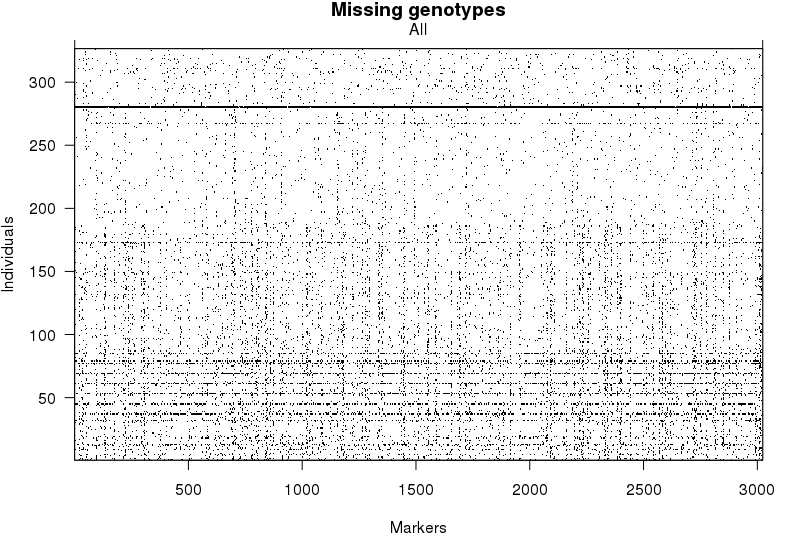
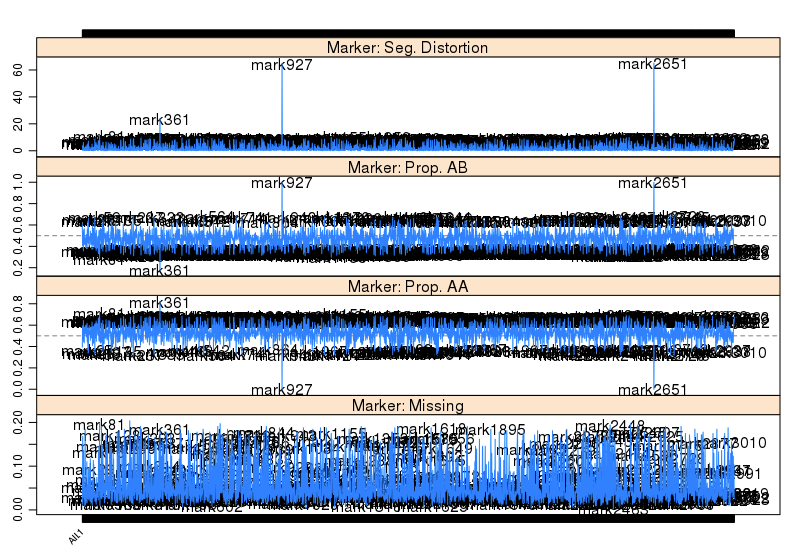
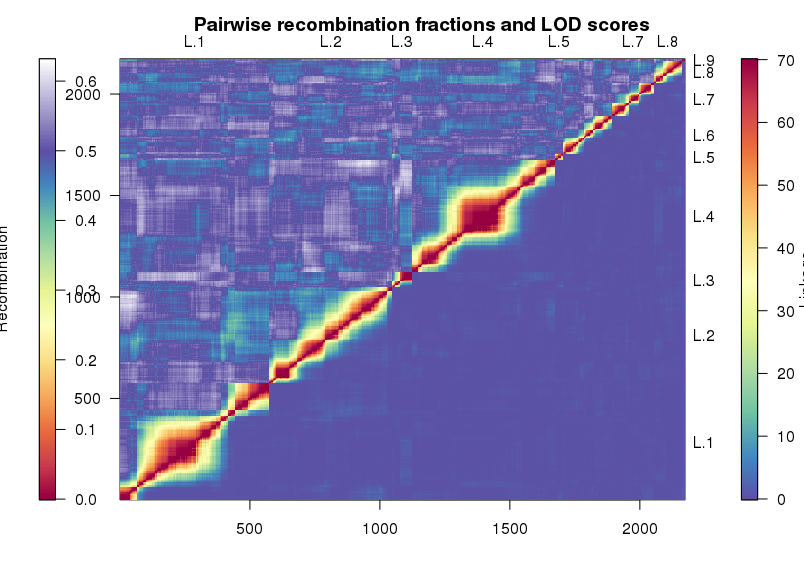
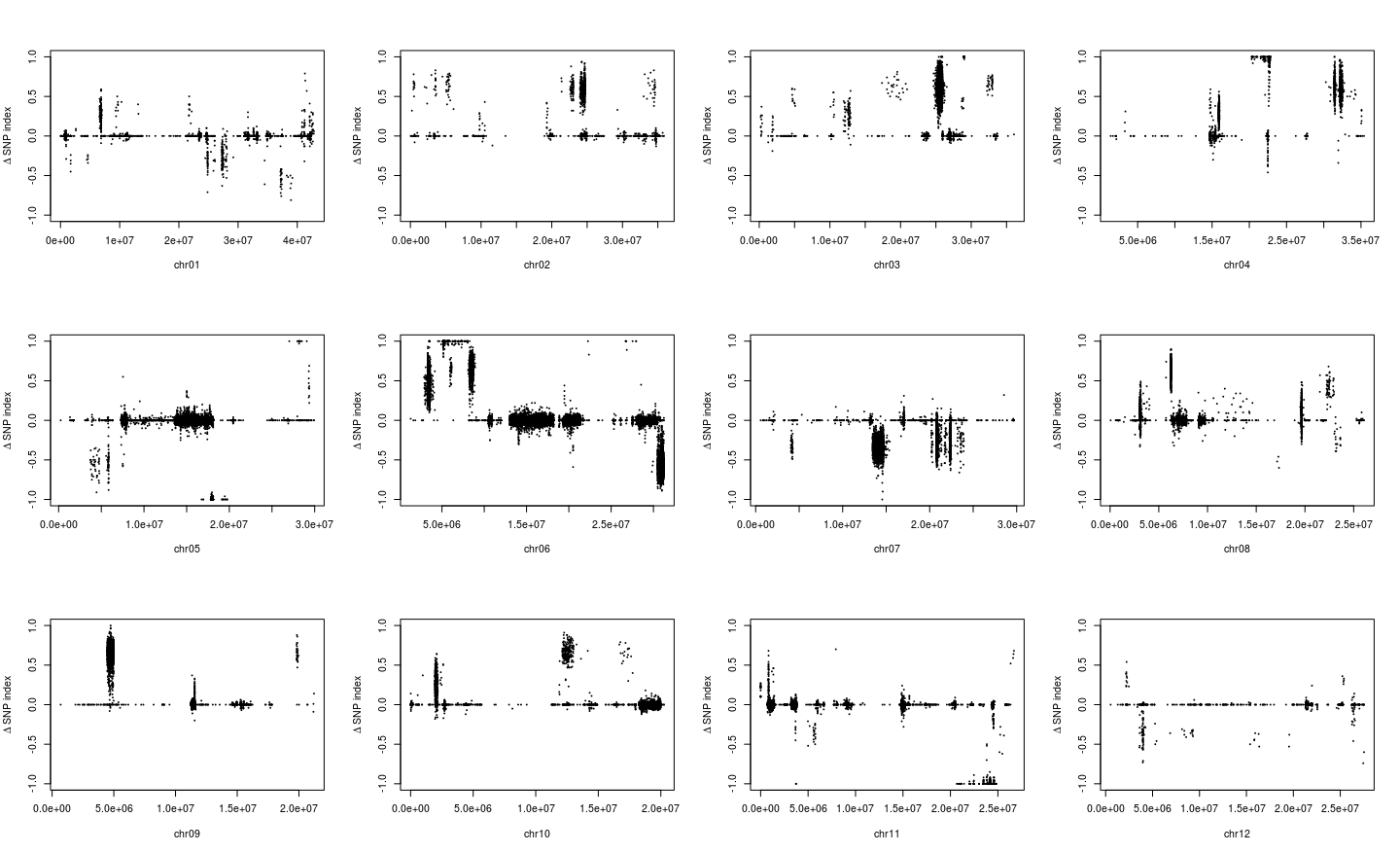
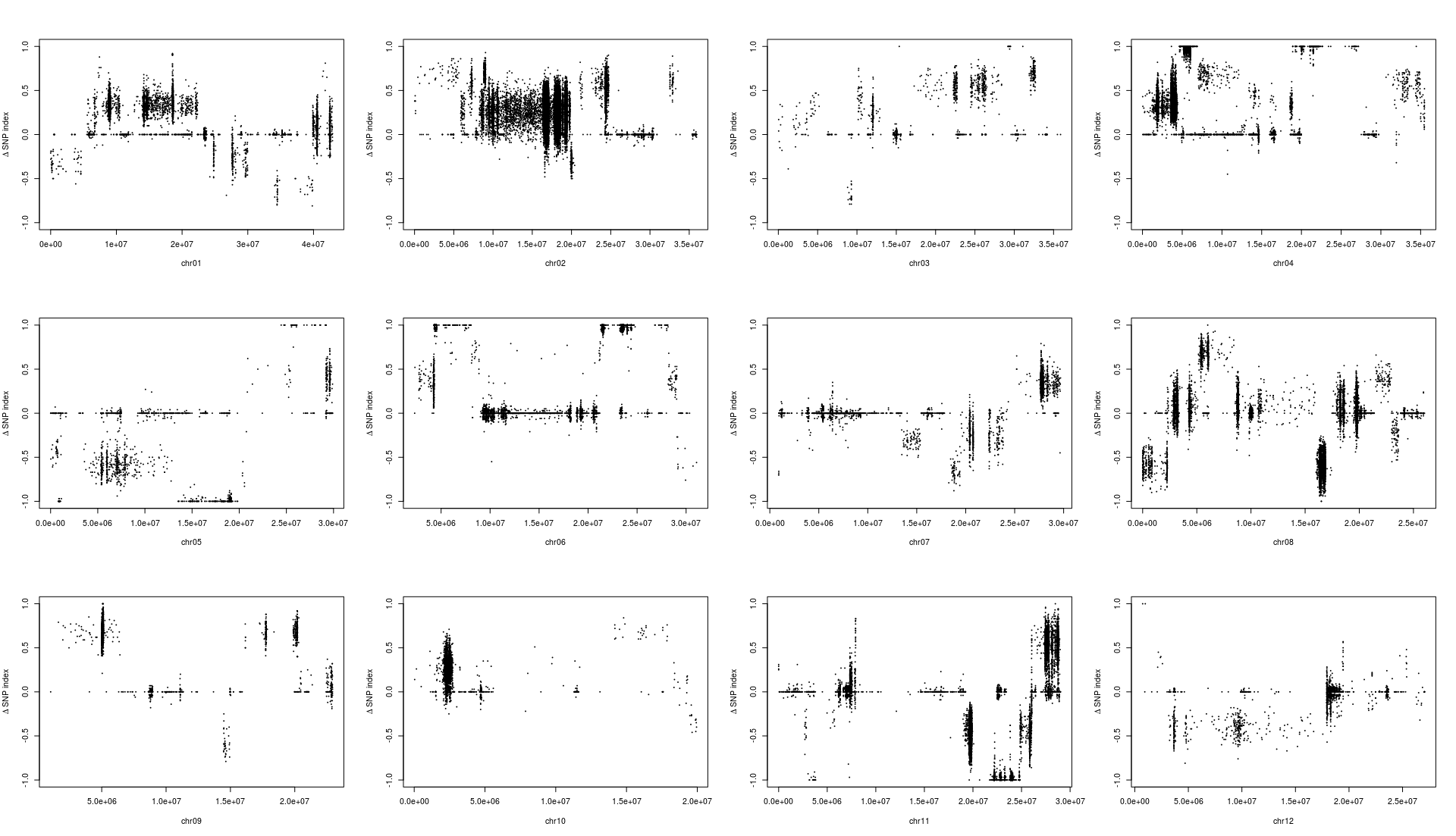
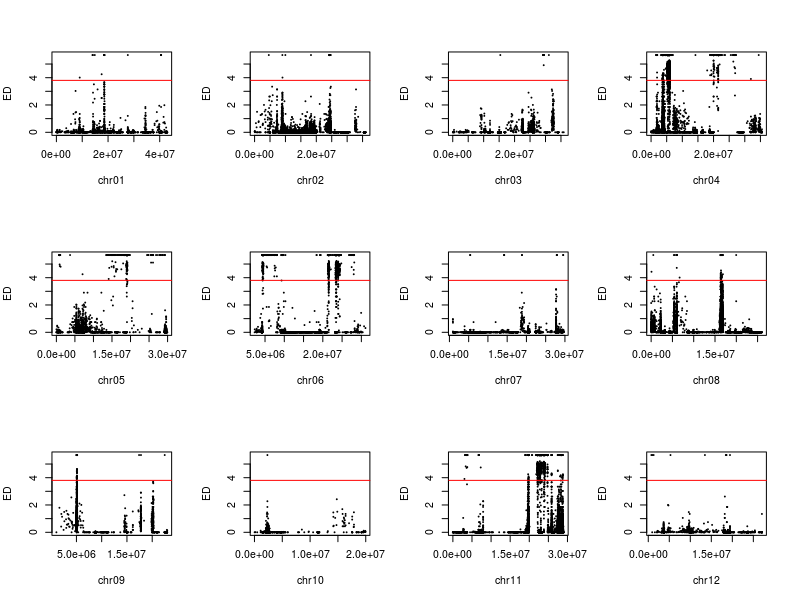
用于讲解的文章题为”Identification of a cold-tolerant locus in rice (Oryza sativa L.) using bulked segregant analysis with a next-generation sequencing strategy”, 发表在Rice上。 文章用的耐寒(Kongyu131)和不耐寒(Dongnong422)的两个亲本进行杂交,得到的F2后代利用单粒传法得到RIL(重组自交系)群体.之后用BWA+GATK识别SNP,计算ED和SNP-index作图。
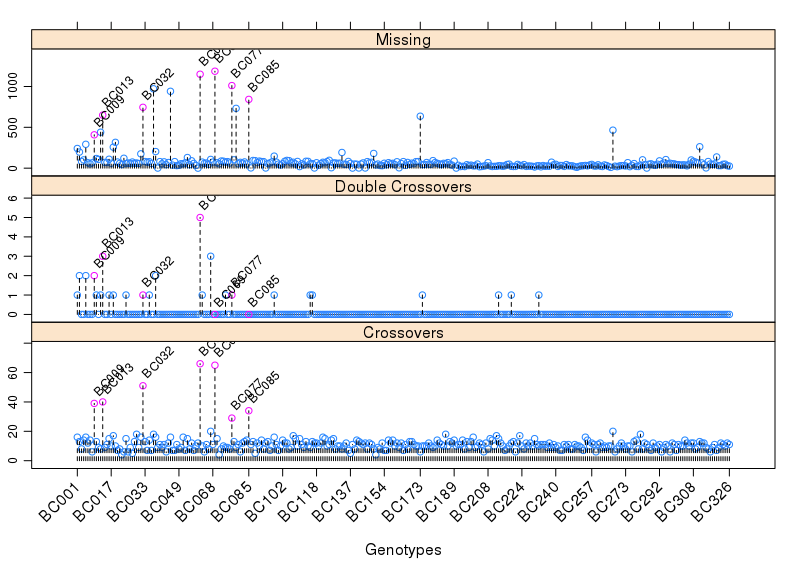
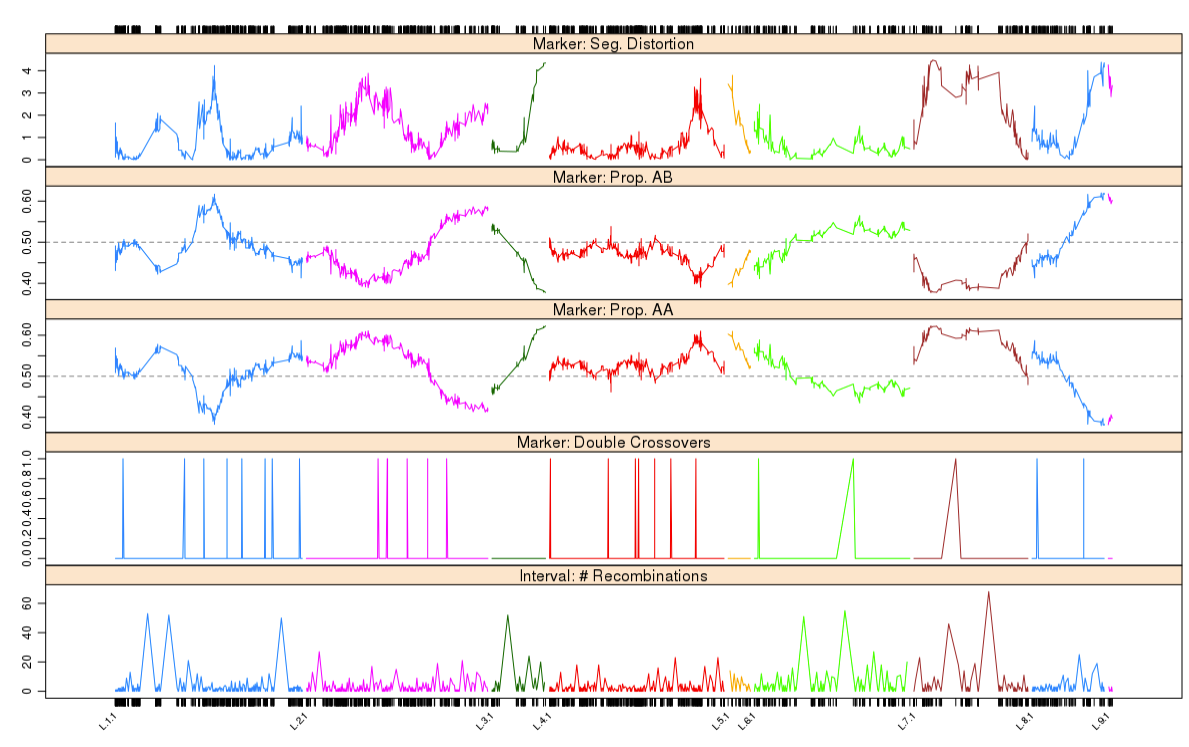
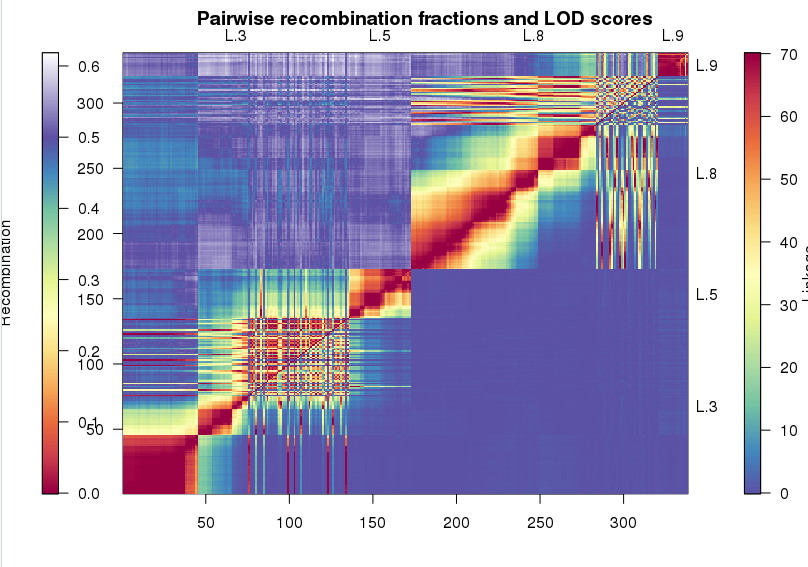
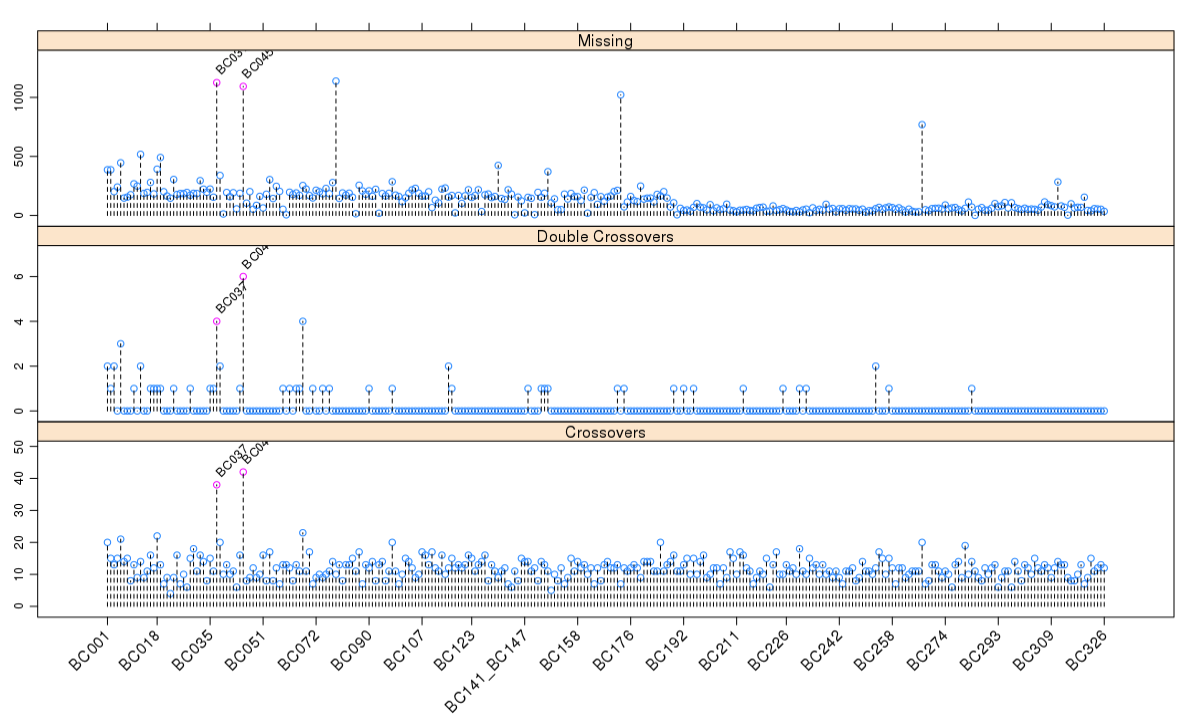
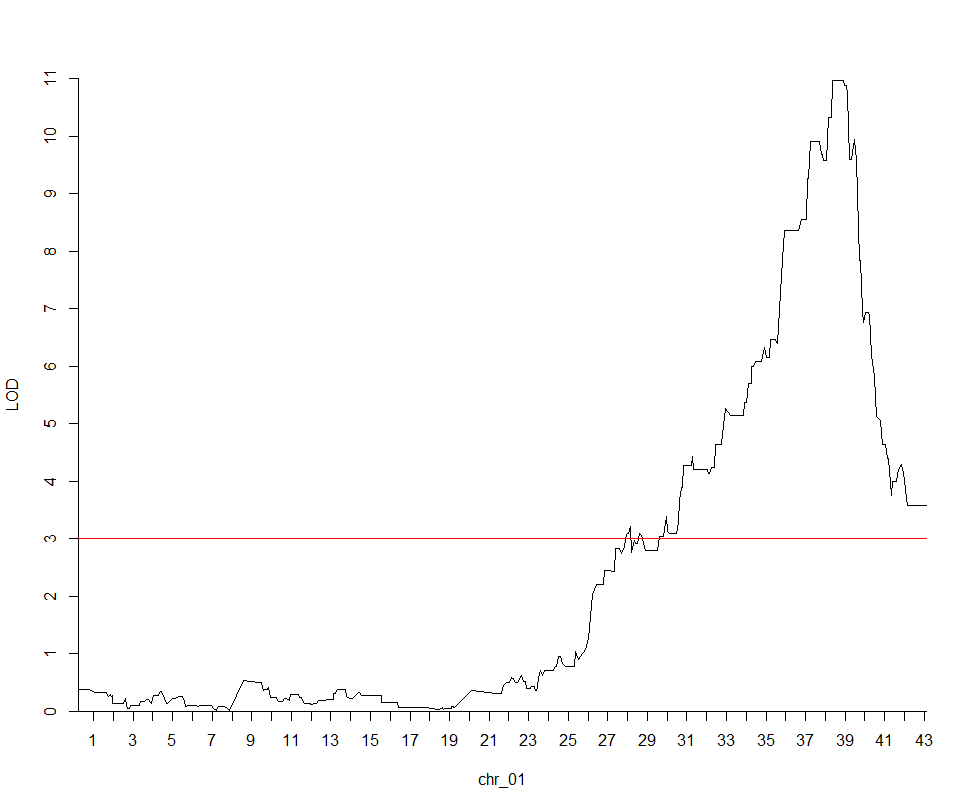
这次实战的目标也就是得到文章关键的两张图:
环境准备
新建一个项目,用于存放本次分析的数据和结果
1 2
mkdir -p rice-bsa cd rice-bsa
后续分析还需要用到如下软件:
wget: 一般Linux系统会自带,用于下载数据
seqkit: 多能的序列处理软件
fastp: 数据质控
bwa: 数据比对到参考基因组
samtools: 处理sam/bam文件
sambamba: 标记重复序列
bcftools: 处理vcf/bcf,能用于变异检测
R: 数据分析
我们需要分别下载参考基因组(IRGSP)数据和测序数据。
1 2 3 4 5 6
# rice-bsa目录下 mkdir -p ref && cd ref wget https://rapdb.dna.affrc.go.jp/download/archive/irgsp1/IRGSP-1.0_genome.fasta.gz gunzip IRGSP-1.0_genome.fasta.gz # 建立索引 bwa index IRGSP-1.0_genome.fasta
LOC_Os06g39740 and LOC_Os06g39750,were annotated as the function of “response to cold (GO: 0009409)”, suggesting their key roles in regulating cold tolerance in rice. “
cd $HOME/biosoft wget http://bioinfo.mpipz.mpg.de/shoremap/SHOREmap_v3.4.tar.gz # 替换SHOREmap下的dislin的一些文件 tar -zxvf SHOREmap_v3,4 rm dislin/*dislin_d.* cp $DISLIN/*dislin_d.* dislin # 编辑/etc/profile或.bashrc vi .bashrc export LD_LIBRARY_PATH=$HOME/src/SHOREmap_v3.4/dislin # 退出保存.bashrc: Esc+:wq source .bashrc # 到之前安装的文件夹下 cd & cd src/SHOREmap_v3.4 (可选,如果没有g++)sudo apt-get install build-essential make # 将编译文件拷贝到习惯的文件夹中,然后添加执行路径 cp SHOREmap ../../biosoft/SHOREmap_v3.4 echo "export $HOME/bisoft/SHOREmap_v3.4" >> ~/.bashrc
最后,可以重新启动一下bash验证
官方网站提供的两个常见问题的解答
Note 1: if the compilation complains like “/usr/bin/ld: cannot find -lXt” (or “/usr/bin/ld: cannot find -ldislin_d”), please open the makefile with the command
1
vi makefile
Press keys ‘esc’ and ‘i’ on the keyboard to edit makefile; move the cursor with arrow keys to the position before -lXt, and edit -L/path/to/libXt.so/; if ‘-ldislin_d’ is not found, edit -L/path/to/dislin_d/ before -ldislin_d). After that, press keys ‘esc’, type in :wq, and press enter to save editing and quit vi. (‘-L’ tells the linker where to find the library given by -l)
Note 2: if ‘/usr/lib/ld: warning: libXm.so.3, needed by ./dislin/libdislin_d.so, not found (try using -rpath or -rpath-link)’ occurs, and you have installed libmotif4, do the following:
1
cp /usr/lib/libXm.so.4 /usr/lib/libXm.so.3
We can make SHOREmap avaiable for general use by inserting the following command into /etc/profile
# -v Output variant sites only (force -c) # -c属于Call variants using Bayesian inference. This option automatically invokes option -e.When -v is in use, skip loci where the fraction of samples covered by reads is below FLOAT. [0],目前被m取代 # -g Call per-sample genotypes at variant sites (force -c),这个没有找到合适的替代参数
# If you are trying to view VCF 4.2 files in IGV - you may run into issues. This function might help you. # This script will: # 1. Rename the file as version 4.1 # 2. Replace parentheses in the INFO lines (IGV doesn't like these!)
function vcf_downgrade() { outfile=${1/.bcf/} outfile=${outfile/.gz/} outfile=${outfile/.vcf/} bcftools view --max-alleles 2 -O v $1 | \ sed "s/##fileformat=VCFv4.2/##fileformat=VCFv4.1/" | \ sed "s/(//" | \ sed "s/)//" | \ sed "s/,Version=\"3\">/>/" | \ bcftools view -O z > ${outfile}.dg.vcf.gz tabix ${outfile}.dg.vcf.gz }
其实对于单个文件而言,可以直接用以下命令
1 2 3 4 5 6 7 8
infile=BG.vcf outfile=BG.vcf bcftools view --max-alleles 2 -O v ${infile} | \ sed "s/##fileformat=VCFv4.2/##fileformat=VCFv4.1/" | \ sed "s/(//" | \ sed "s/)//" | \ sed "s/,Version=\"3\">/>/" | \ bcftools view -O z > ${outfile}.dg.vcf.gz
使用SHOREmap寻找突变所在区
第一步:需要把bcf文件通过SHOREmap convert转换成SHOREmap能认识的格式
1
SHOREmap convert --marker samtools.vcf --folder path/to/folder -runid int
会生成三个文件3_converted_consen.txt, 3_converted_variant.txt and 3_converted_reference.txt.
候选marker文件。也就是使用SHOREmap convert通过vcf生成的converted_variant.txt,每一列的含义如下。 Column Description 1 Project name 2 Identity of chromosome 3 Position of the SNP-marker 4 Reference base 5 Alternative base (or mutant base) 6 Quality of the alternative base (ranging from 0 to 40) 7 Number of reads supporting the predicted base 8 Ratio of reads supporting the predicted base to total coverage
wget https://mirrors.tuna.tsinghua.edu.cn/CRAN/src/base/R-3/R-3.6.1.tar.gz tar -zxvf R-3.6.1.tar.gz cd R-3.6.1 # --enable-R-shlib for Rstudio server mkdir -p /opt/sysoft ./configure --enable-R-shlib --prefix=/opt/sysoft/R-3.6.1 make -j 8 make install
R 3.4 packages for Ubuntu on i386 and amd64 are available for all stable Desktop releases of Ubuntu prior to Bionic Beaver (18.04) until their official end of life date.
deb https://cloud.r-project.org/bin/linux/ubuntu disco-cran35/ deb https://cloud.r-project.org/bin/linux/ubuntu cosmic-cran35/ deb https://cloud.r-project.org/bin/linux/ubuntu bionic-cran35/ deb https://cloud.r-project.org/bin/linux/ubuntu xenial-cran35/ deb https://cloud.r-project.org/bin/linux/ubuntu trusty-cran35/
wget https://mirrors.tuna.tsinghua.edu.cn/CRAN/src/base/R-3/R-3.6.1.tar.gz tar -zxvf R-3.6.1.tar.gz cd R-3.6.1 # --enable-R-shlib for Rstudio server ./configure --enable-R-shlib --prefix=/opt/sysoft/R-3.6.1 make -j 8 make install
不同平台有着不同的Make工具用于编译,例如 GNU Make ,QT 的 qmake ,微软的 MS nmake,BSD Make(pmake),Makepp,等等。这些 Make 工具遵循着不同的规范和标准,所执行的 Makefile 格式也千差万别。这样就带来了一个严峻的问题:如果软件想跨平台,必须要保证能够在不同平台编译。而如果使用上面的 Make 工具,就得为每一种标准写一次 Makefile ,这将是一件让人抓狂的工作。
CMake就是针对上面问题所设计的工具:它首先允许开发者编写一种平台无关的 CMakeList.txt 文件来定制整个编译流程,然后再根据目标用户的平台进一步生成所需的本地化 Makefile 和工程文件,如 Unix 的 Makefile 或 Windows 的 Visual Studio 工程。从而做到“Write once, run everywhere”。显然,CMake 是一个比上述几种 make 更高级的编译配置工具。一些使用 CMake 作为项目架构系统的知名开源项目有 VTK、ITK、KDE、OpenCV、OSG 等.
1 2 3
wget https://cmake.org/files/v3.10/cmake-3.10.2.tar.gz tar xf cmake-3.10.2.tar.gz cd cmake-3.10.2
wget ftp://ftp.invisible-island.net/ncurses/ncurses.tar.gz && tar -zxvf ncurses.tar.gz ./configure --enable-shared --prefix=$HOME/usr make && make install
# libevent cd src wget https://github.com/libevent/libevent/releases/download/release-2.1.8-stable/libevent-2.1.8-stable.tar.gz tar -zxvf libevent-2.1.8-stable.tar.gz && cd libevent-2.1.8 ./configure prefix=$HOME/usr && make && make install
bzip2, xz, zlib: 文件压缩相关函数库,后续samtools编译时需要。
1 2 3 4 5 6
wget http://www.zlib.net/zlib-1.2.11.tar.gz tar -zxvf zlib-1.2.11.tar.gz && cd zlib-1.2.11 && ./configure --prefix=$HOME/usr && make && make install wget http://www.bzip.org/1.0.6/bzip2-1.0.6.tar.gz tar -zxvf bzip2-1.0.6.tar.gz && cd bzip2-1.0.6 && ./configure --prefix=$HOME/usr && make && make install wget https://tukaani.org/xz/xz-5.2.3.tar.gz tar -zxvf xz-5.2.3.tar.gz && cd xz-5.2.3 && ./configure --prefix=$HOME/usr && make && make install
wget http://ftp.gnu.org/gnu/readline/readline-7.0.tar.gz tar -zxvf readline-7.0.tar.gz && cd readline-7.0 ./configure --prefix=$HOME/usr && make && make install
PCRE: 提供和Perl5相同语法和语义正则表达式的函数库,后续安装R用到。
1 2 3
wget ftp://ftp.csx.cam.ac.uk/pub/software/programming/pcre/pcre-8.41.tar.gz tar -zxvf pcre-8.41.tar.gz && cd pcre-8.41 ./configure --enable-utf --enable-pcregrep-libz --enable-pcregrep-libbz2 --prefix=$HOME/usr
# 安装X11 wget -4 https://www.x.org/releases/X11R7.7/src/lib/libX11-1.5.0.tar.gz tar -zxvf libX11-1.5.0.tar.gz && cd libX11-1.5.0 ./configure --prefix=$HOME/usr && make && make install
wget -O zsh.tar.gz https://sourceforge.net/projects/zsh/files/latest/download tar -zxvf zsh.tar.gz && cd zsh export CPPFLAGS="-I$HOME/usr/include/" LDFLAGS="-L$HOME/usr/lib" ../configure --prefix=$HOME/usr --enable-shared make && make install
export CPPFLAGS="-I$HOME/usr/include -I$HOME/usr/include/ncurses" export LDFLAGS="-L$HOME/usr/lib -L$HOME/usr/lib64" mkdir -p src && cd src git clone https://github.com/tmux/tmux.git cd tmux sh autogen.sh ./configure --prefix=$HOME/usr make && make install
这里选择了mpi=1.0=mpich, 原因是i-adhore依赖于mpich. 如果安装了openmpi就会出现error while loading shared libraries: libmpi_cxx.so.40: cannot open shared object file: No such file or directory
tar -zxvf i-adhore-3.0.01.tar.gz cd i-adhore-3.0.01 mkdir -p build && cd build cmake .. -DCMAKE_INSTALL_PREFIX=~/opt/miniconda3/envs/wgd/ make -j 4 make insatall
Tang, H., Bowers, J.E., Wang, X., Ming, R., Alam, M., and Paterson, A.H. (2008). Synteny and Collinearity in Plant Genomes. Science 320, 486–488.
Tiley, G.P., Barker, M.S., and Burleigh, J.G. (2018). Assessing the Performance of Ks Plots for Detecting Ancient Whole Genome Duplications. Genome Biol Evol 10, 2882–2898.
Guo, L., Winzer, T., Yang, X., Li, Y., Ning, Z., He, Z., Teodor, R., Lu, Y., Bowser, T.A., Graham, I.A., et al. (2018). The opium poppy genome and morphinan production. Science 362, 343–347.
Zwaenepoel, A., and Van de Peer, Y. (2019). wgd—simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics 35, 2153–2155.
config_version: 0.2 genome_folder: /path/to/reference genome_server: http://refgenomes.databio.org genomes: TAIR10: assets: fasta: path: fasta/TAIR10.fa asset_description: Sequences in the FASTA format fai: path: fasta/TAIR10.fa.fai asset_description: Indexed fasta file, produced with samtools faidx chrom_sizes: path: fasta/TAIR10.chrom.sizes asset_description: Chromosome sizes file bwa_index: path: bwa_index asset_description: Genome index for Burrows-Wheeler Alignment Tool, produced with bwa index