BSA虽然不是我最早接触的高通量数据分析项目(最早的是RNA-seq),但是却是我最早独立开展的数据分析项目, 我曾经专门写过一篇文章介绍如何使用SHOREMap做拟南芥的EMS诱变群体的BSA分析
在遗传定位上,相对于GWAS和binmap,BSA是一个比较省钱的策略,它只需要测两个亲本和后代中两个极端差异群体即可,但是它对实验设计,表型考察,样本挑选都有比较高的要求。如果你的表型差异并不是泾渭分明,那么还是不要用BSA比较合适。这方面知识,建议阅读徐云碧老师2016年发表在PBJ上的”Bulked sample analysis in genetics, genomics and crop improvement”, 这篇教程侧重于实际分析,而非理论讲解。
用于讲解的文章题为”Identification of a cold-tolerant locus in rice (Oryza sativa L.) using bulked segregant analysis with a next-generation sequencing strategy”, 发表在Rice上。 文章用的耐寒(Kongyu131)和不耐寒(Dongnong422)的两个亲本进行杂交,得到的F2后代利用单粒传法得到RIL(重组自交系)群体.之后用BWA+GATK识别SNP,计算ED和SNP-index作图。
这次实战的目标也就是得到文章关键的两张图:
环境准备 新建一个项目,用于存放本次分析的数据和结果
1 2 mkdir -p rice-bsa cd rice-bsa
后续分析还需要用到如下软件:
wget: 一般Linux系统会自带,用于下载数据
seqkit: 多能的序列处理软件
fastp: 数据质控
bwa: 数据比对到参考基因组
samtools: 处理sam/bam文件
sambamba: 标记重复序列
bcftools: 处理vcf/bcf,能用于变异检测
R: 数据分析
我们需要分别下载参考基因组(IRGSP)数据和测序数据。
1 2 3 4 5 6 mkdir -p ref && cd refwget https://rapdb.dna.affrc.go.jp/download/archive/irgsp1/IRGSP-1.0_genome.fasta.gz gunzip IRGSP-1.0_genome.fasta.gz bwa index IRGSP-1.0_genome.fasta
之后 根据文章提供的编号,SRR6327815, SRR6327816, SRR6327817, SRR6327818 ,我们到ENA 查找对应的下载链接进行数据下载。
1 2 3 4 5 6 mkdir -p data && cd datawget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR632/005/SRR6327815/SRR6327815_{1,2}.fastq.gz wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR632/006/SRR6327816/SRR6327816_{1,2}.fastq.gz wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR632/007/SRR6327817/SRR6327817_{1,2}.fastq.gz wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR632/008/SRR6327818/SRR6327818_{1,2}.fastq.gz
因为在NCBI上下载的是SRA格式,需要做数据转换,而从ENA上可以直接下载压缩过的fastq文件,所以我更偏好于ENA。
上游预处理 对于测序数据而言,为了能够获得后续用于计算SNP-index或者ED的SNP信息,我们需要将二代测序得到的数据回贴到参考基因组上,然后利用变异检测软件找到每个样本和参考基因组的区别。
数据质控 原始数据中可能有一部分序列质量不够高,会影响后续分析,比如说测序质量不够,或者说存在接头。通常我们会都会对原始数据进行一波过滤,这里用的是fastp,优点就是快。
1 2 3 4 5 6 mkdir -p 01-clean-datafastp -i data/SRR6327815_1.fastq.gz -I data/SRR6327815_2.fastq.gz -o 01-clean-data/SRR6327815_1.fastq.gz -O 01-clean-data/SRR6327815_2.fastq.gz fastp -i data/SRR6327816_1.fastq.gz -I data/SRR6327816_2.fastq.gz -o 01-clean-data/SRR6327816_1.fastq.gz -O 01-clean-data/SRR6327816_2.fastq.gz fastp -i data/SRR6327817_1.fastq.gz -I data/SRR6327817_2.fastq.gz -o 01-clean-data/SRR6327817_1.fastq.gz -O 01-clean-data/SRR6327817_2.fastq.gz fastp -i data/SRR6327818_1.fastq.gz -I data/SRR6327818_2.fastq.gz -o 01-clean-data/SRR6327818_1.fastq.gz -O 01-clean-data/SRR6327818_2.fastq.gz
序列比对 之后将各个样本的序列回帖到参考基因组
1 2 3 4 5 6 7 8 9 10 11 mkdir -p 02-read-alignNUMBER_THREADS=80 REFERENCE=ref/IRGSP-1.0_genome.fasta for i in `ls 01-clean-data/`; do sample=${i%%_*} (bwa mem -M -R "@RG\\tID:${sample} \\tSM:${sample} \\tPL:ILLUMINA" \ -t $NUMBER_THREADS $REFERENCE 01-clean-data/${sample} _1.fastq.gz 01-clean-data/${sample} _2.fastq.gz || echo -n 'error' ) \ | samtools sort -@ 20 -o 02-read-align/${sample} _sort.bam - samtools index -@ 20 02-read-align/${sample} _sort.bam done
这里用到了稍微比较高级的shell操作
变量名替换sample=${i%%_*}
逻辑判断: ||
子shell: ()
for循环: for do done
接着我门需要去除重复序列,这里的重复序列指的是拥有相同位置信息,且序列也一模一样的read,通常是由PCR扩增引起。过滤重复序列的目的是为了提高变异检测的准确性,如果一条read上出现的“变异”其实是在第一轮PCR扩增时引入的错误,那么后续的扩增只会让这个错误一直保留着,随后测序的时候这条许多又被多次测到,那么在后续的分析中由于多次出现,就有可能会变异检测软件当作真实变异。
这里用sambamba,因为它的速度比较快,且结果和picard一模一样。
1 2 3 4 5 for i in `ls 02-read-align/*_sort.bam`; do sample=${i%%_*} sambamba markdup -r -t 10 ${sample} _sort.bam ${sample} _mkdup.bam done
变异检测 变异检测最常见的就是bcftools, freebayes和GATK. 这里用的是BCFtools,主要原因还是它的速度比较快。
这里为了让他的速度更快,我用了--region参数分染色体并行,由于水稻有12条染色体,相当于提速了12倍
1 2 3 4 5 6 7 8 9 10 11 12 13 14 mkdir -p 03-variantsls -1 02-read-align/*_mkdup.bam > bam_list.txtseqkit seq -n ref/IRGSP-1.0_genome.fasta | \ while read regiondo bcftools mpileup -f ref/IRGSP-1.0_genome.fasta \ --redo-BAQ --min-BQ 30 \ --per-sample-mF \ --annotate FORMAT/AD,FORMAT/DP \ --regions ${region} \ -Ou --bam-list bam_list.txt | \ bcftools call -mv -Ob -o 03-variants/${region} .bcf & done
之后将拆分运行的结果合并到单个文件中
1 2 3 mkdir -p 04-variant-filterbcftools concat --naive -o 04-variant-filter/merged.bcf 03-variants/*.bcf
变异过滤 得到VCF文件还需要进行一些过滤,来提高变异的准确性. 这个需要根据具体的项目来进行, 但是有一些固定的指标可以用来对结果进行过滤,例如
测序深度
非参考基因组的高质量read数
是否和indel紧邻,通常和indel比较近的snp都不可靠
平均的比对质量,
举个例子:
1 2 3 4 cd 04-variant-filterbcftools filter -g3 -G10 -e'%QUAL<10 || (RPB<0.1 && %QUAL<15) || (AC<2 && %QUAL<15) || MQ < 30 || MQSB <=0.1' merged.bcf > filter.vcf
之后,我只选择了snp用于后续分析
1 2 bcftools view -i 'TYPE="snp" & N_ALT =1 & STRLEN(ALT) = 1' filter.vcf > snps.vcf
最终留下了将近80w的SNP。这就是后续R语言下游分析的基础。
上游处理后的数据可以从链接:https://pan.baidu.com/s/1n6CX33E6Qrpf85INMxGGAQ ( 密码:q9u3 )下载
下游分析 下游分析可能是比较成熟化的流程分析,对服务器的要求比较高一些,下游分析则是对背景知识要求高一些,例如VCF的格式,遗传学的三大定律等。
让我们先安装并加载所需的R包
1 2 3 4 5 install.pacakges( "devtools" ) install.packages( "vcfR" ) devtools: install_github( "xuzhougeng/binmapr" ) library( "vcfR" ) library( "binmapr" )
然后我们需要利用vcfR读取VCF文件
1 vcf <- read.vcfR( "04-variant-filter/snps.vcf" )
接着从VCF对象中提取两个关键信息,AD(Allele Depth)和GT(Genotype)
1 2 gt <- extract.gt( vcf) ad <- extract.gt( vcf, "AD" )
gt是一个基因型矩阵,基于之的前过滤操作,所以这里只会有”0/0”, “0/1”,”1/1”这三种情况,而ad则是等位基因的count数. 我们用head查看前10行来了解下情况,
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 > head( gt) SRR6327815 SRR6327816 SRR6327817 SRR6327818 chr01_1151 "0/0" "1/1" "0/0" "0/1" chr01_6918 "0/0" "1/1" "0/0" "0/1" chr01_17263 "0/0" "1/1" "0/0" "0/1" chr01_21546 "1/1" "1/1" "0/0" "0/1" chr01_24732 "1/1" "0/0" "0/0" "0/0" chr01_33667 "1/1" "1/1" "0/0" "0/1" > head( ad) SRR6327815 SRR6327816 SRR6327817 SRR6327818 chr01_1151 "14,0" "0,18" "10,0" "11,3" chr01_6918 "31,0" "0,30" "32,0" "21,5" chr01_17263 "46,0" "0,34" "20,0" "21,8" chr01_21546 "0,25" "0,20" "17,0" "16,3" chr01_24732 "0,29" "36,0" "21,0" "31,0" chr01_33667 "0,26" "0,31" "28,0" "29,11"
其中SRR6327815, SRR6327816, SRR6327817, SRR6327818 分别对应着 KY131, DN422, T-pool 和 S-pool
仔细观察的话,你会发现一个chr01_21546是一个有趣的位置,因为双亲都是纯合情况下,SRR6327818居然是 “16,3”, 基因型是”0/1”, 这既有可能是亲本是杂合但没有测到,也有可能是后代测错了,一个简单粗暴的方法就是删掉它。此外我们还需要考虑是选择KY131是”1/1”且 “DN422” 是 “0/0”的位点。还是选择KY131是”0/0”, 且 “DN422” 是 “1/1”位点。最好的方法就是两种都测试一下。
首先测试KY131是”1/1”且 “DN422” 是 “0/0”的位点
1 2 3 4 mask <- which( gt[ , "SRR6327815" ] == "1/1" & gt[ , "SRR6327816" ] == "0/0" ) ad_flt <- ad[ mask, c ( "SRR6327817" , "SRR6327818" ) ] colnames( ad_flt) <- c ( "T_Pool" , "S_Pool" )
这一波过滤,我们从原来的80w位点中留下了20w个位点,因此测双亲很重要 ,能够极大的降低噪音。
然后,我们就可以根据AD计算SNP-index,即 ALT_COUNT / (REF_COUNT + ALT_COUNT), 在0-1之间。
1 freq <- calcFreqFromAd( ad_flt, min.depth = 10 , max.depth = 100 )
这里设了一个最大和最小深度用于计算频率,太大的深度可能是同源基因或者是重复序列,太低的深度在计算的时候不太准确。
1 freq2 <- freq[ Matrix:: rowSums( is.na ( freq) ) == 0 , ]
接着我们就可以尝试去重现文章的结果了
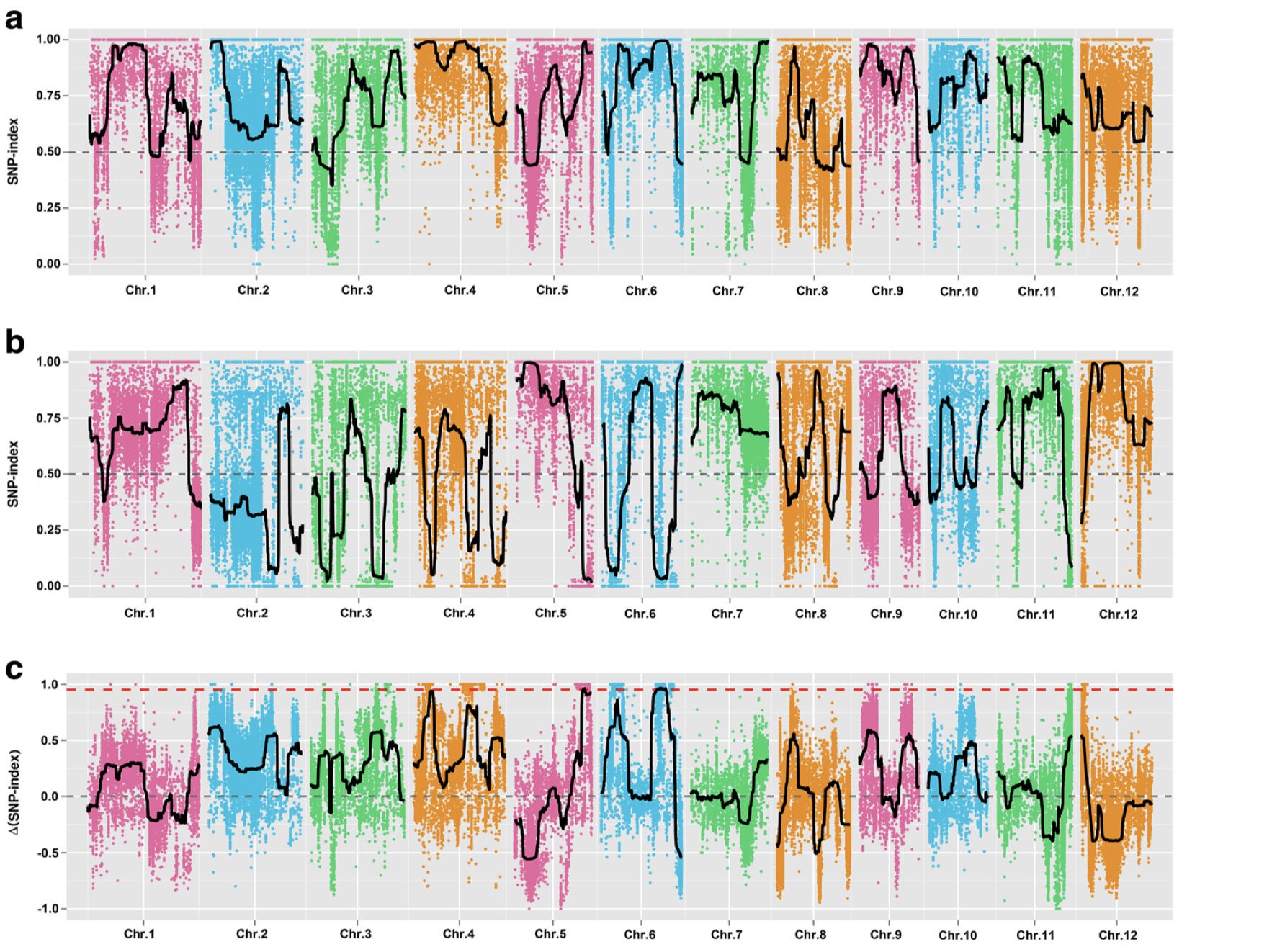
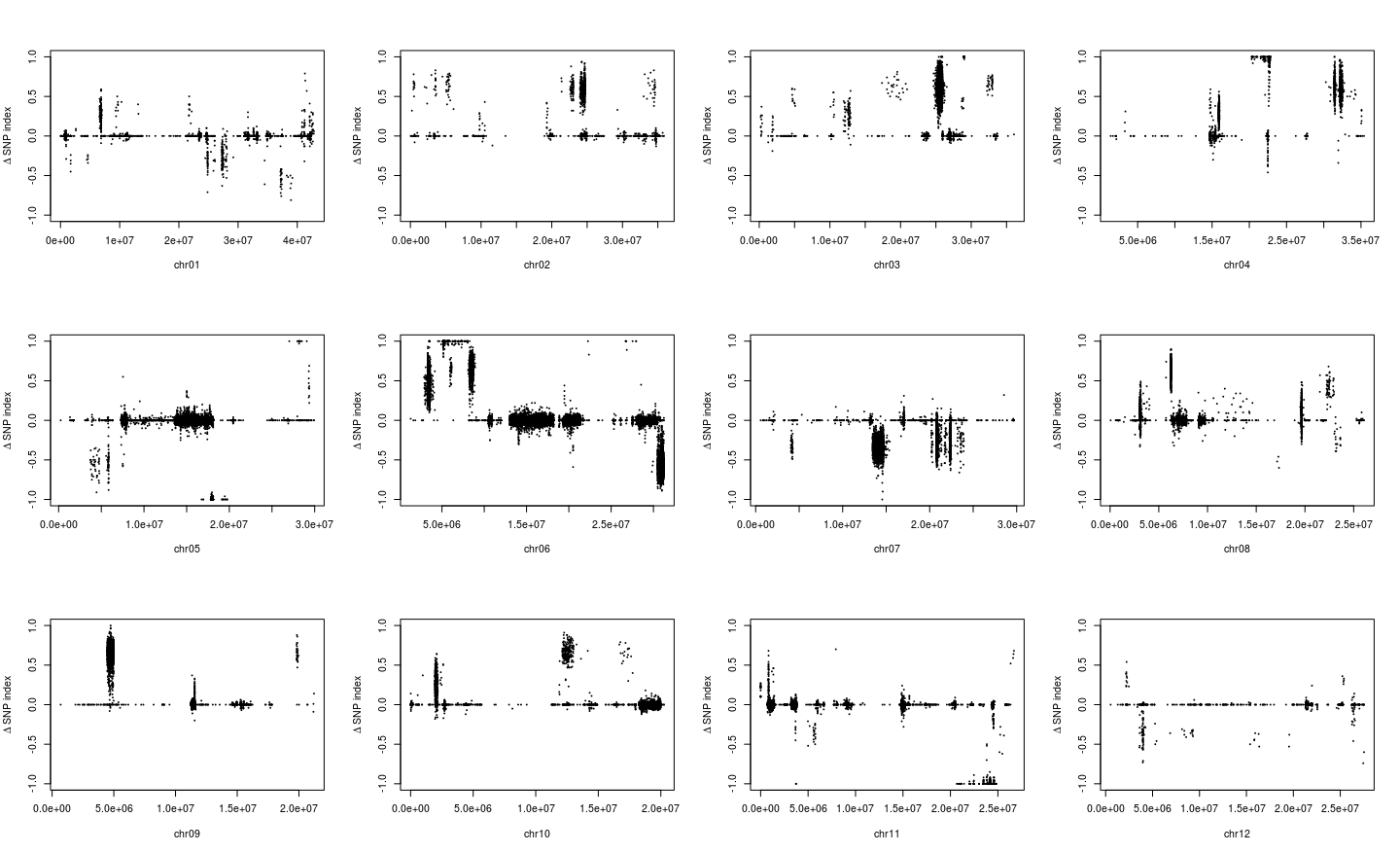
1 2 3 4 5 6 7 8 9 10 par( mfrow = c ( 3 , 4 ) ) for ( i in paste0( "chr" , formatC( 1 : 12 , width = 2 , flag= 0 ) ) ) { freq_flt <- freq2[ grepl( i, row.names( freq2) ) , ] pos <- as.numeric ( substring( row.names( freq_flt) , 7 ) ) plot( pos, freq_flt[ , 2 ] - freq_flt[ , 1 ] , ylim = c ( - 1 , 1 ) , pch = 20 , cex = 0.2 , xlab = i, ylab = expression ( paste( Delta, " " , "SNP index" ) ) ) }
从图中,我惊讶的发现一些染色体上的部分区域居然都没有标记了,所以我们选择KY131是”0/0”, 而 “DN422” 是 “1/1”的位点进行分析
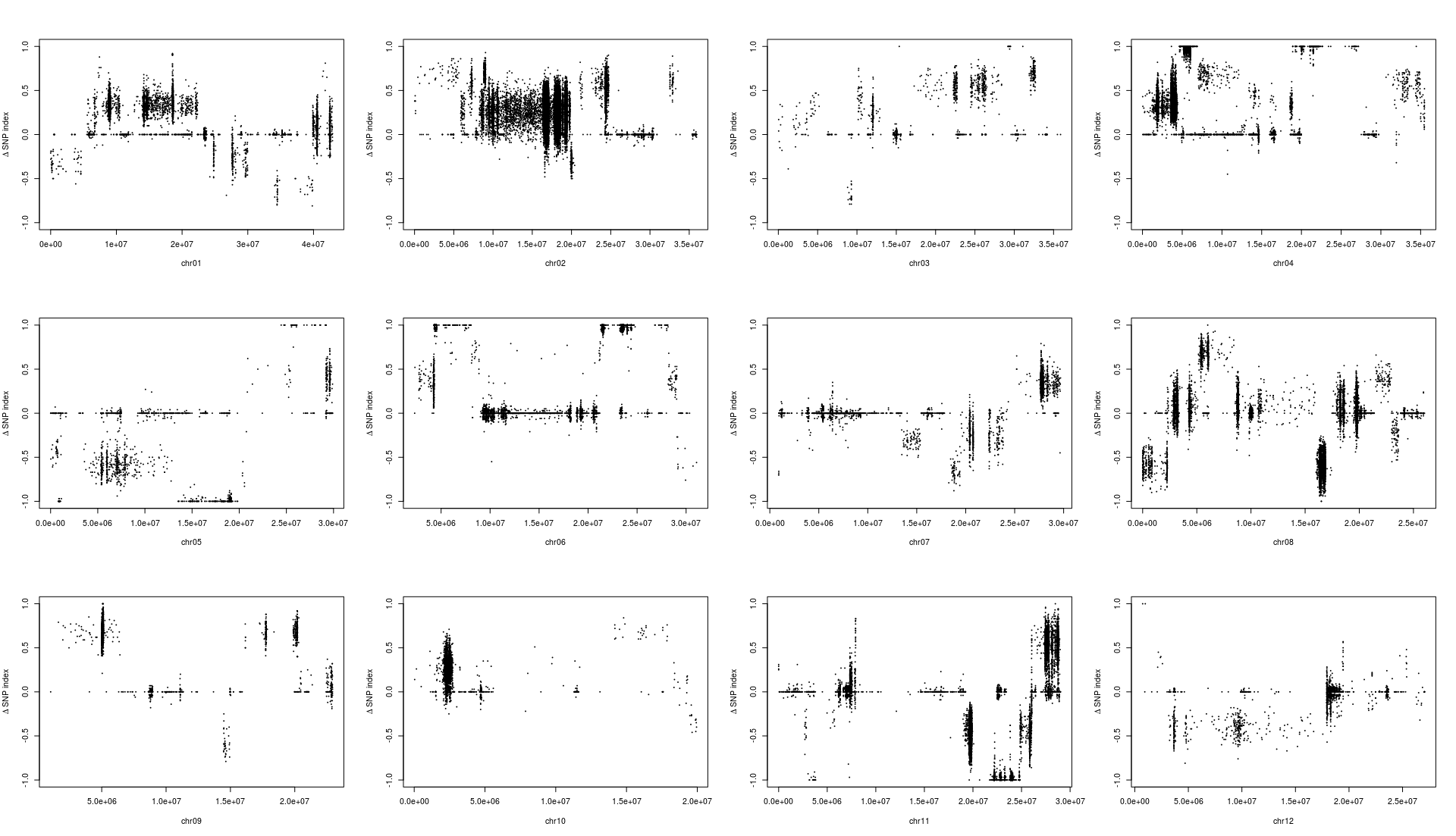
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 mask <- which( gt[ , "SRR6327815" ] == "0/0" & gt[ , "SRR6327816" ] == "1/1" ) ad_flt <- ad[ mask, c ( "SRR6327817" , "SRR6327818" ) ] colnames( ad_flt) <- c ( "T_Pool" , "S_Pool" ) freq <- calcFreqFromAd( ad_flt, min.depth = 10 , max.depth = 100 ) freq2 <- freq[ Matrix:: rowSums( is.na ( freq) ) == 0 , ] par( mfrow = c ( 3 , 4 ) ) for ( i in paste0( "chr" , formatC( 1 : 12 , width = 2 , flag= 0 ) ) ) { freq_flt <- freq2[ grepl( i, row.names( freq2) ) , ] pos <- as.numeric ( substring( row.names( freq_flt) , 7 ) ) plot( pos, freq_flt[ , 1 ] - freq_flt[ , 2 ] , ylim = c ( - 1 , 1 ) , pch = 20 , cex = 0.2 , xlab = i, ylab = expression ( paste( Delta, " " , "SNP index" ) ) ) }
从结果中,我们能够发现一个有趣的结果,不同的筛选标记标准会导致标记在染色体上分布发生变化,这可能意味着在某种的筛选标准下,会使得和性状有关的位点明显的被过滤掉。
根据文章里的结果,候选基因落在6号染色体的20-25M中,也就是选择KY131是”0/0”, 而 “DN422” 是 “1/1”的位点结果和原文比较类似。那么如果我们原本不知道这个结果应该怎么办?这其实也不是什么问题,像我这样把两幅图都做出来,然后和实验设计者交流下,也就差不多知道答案了。
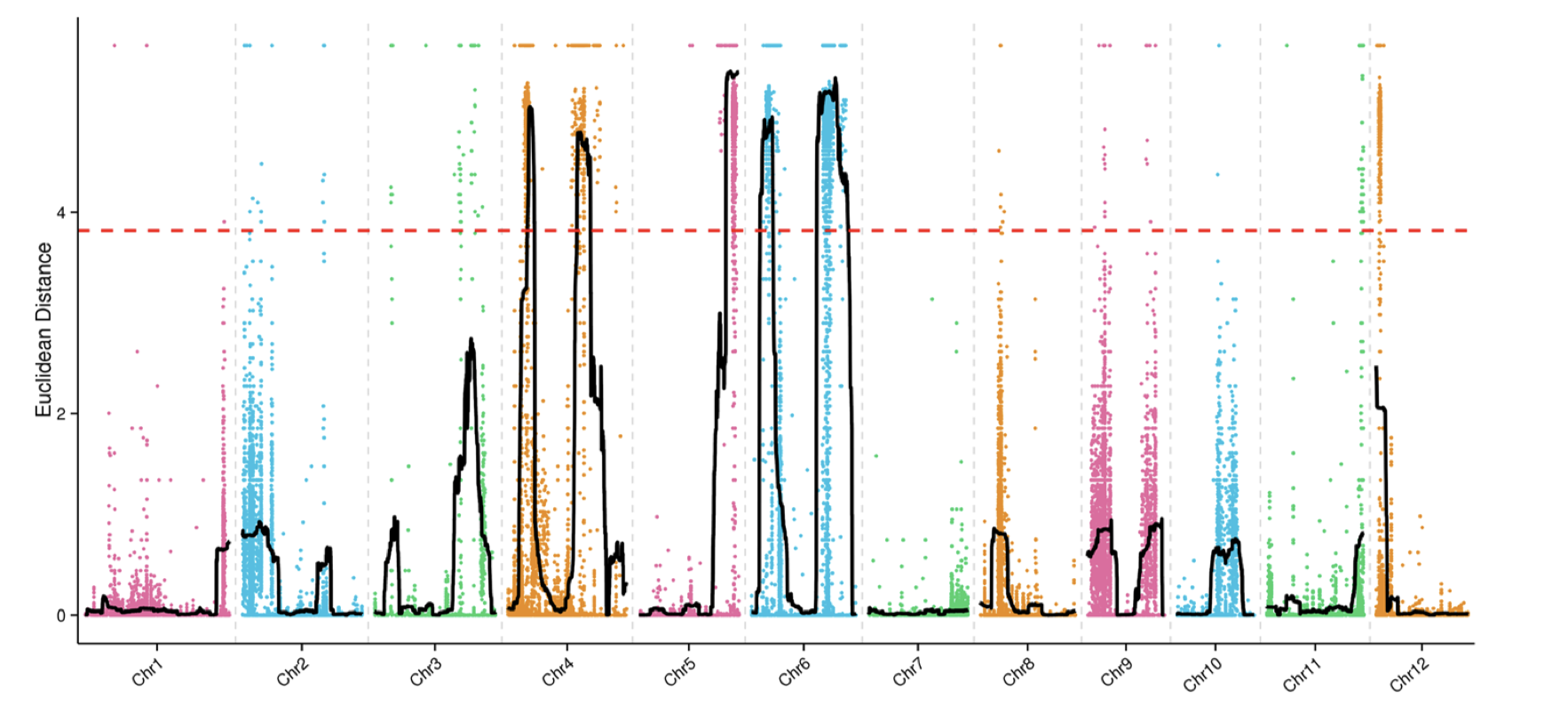
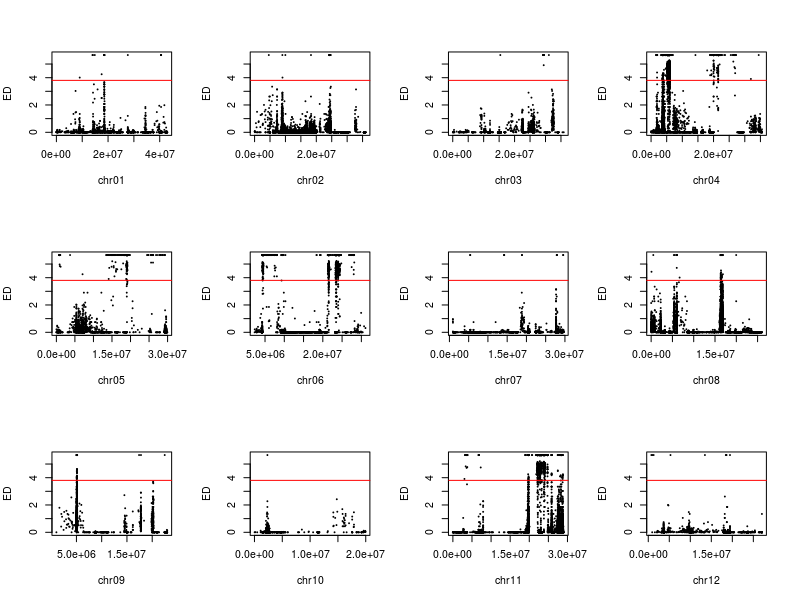
除了SNP-index外,文章还有一个ED(Euclidean distance)方法用于定位,我根据文章的公式和自己的理解写了下代码
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 mask <- which( gt[ , "SRR6327815" ] == "0/0" & gt[ , "SRR6327816" ] == "1/1" ) ad_flt <- ad[ mask, c ( "SRR6327817" , "SRR6327818" ) ] ED_list <- apply( ad_flt, 1 , function ( x) { count <- as.numeric ( unlist( strsplit( x, "," , fixed = TRUE , useBytes = TRUE ) ) ) depth1 <- count[ 1 ] + count[ 2 ] depth2 <- count[ 3 ] + count[ 4 ] ED <- sqrt ( ( count[ 3 ] / depth2 - count[ 1 ] / depth1) ^ 2 + ( count[ 4 ] / depth2- count[ 2 ] / depth1) ^ 2 ) return ( ED^ 5 ) } ) par( mfrow = c ( 3 , 4 ) ) for ( i in paste0( "chr" , formatC( 1 : 12 , width = 2 , flag= 0 ) ) ) { ED_flt <- ED_list[ grepl( i, names ( ED_list) ) ] pos <- as.numeric ( substring( names ( ED_flt) , 7 ) ) plot( pos, ED_flt, pch = 20 , cex = 0.2 , xlab = i, ylab = "ED" ) }
我复现的结果和文章的区别在于,少一个拟合线,也就是以1Mb为区间,每次滑动10Kb计算均值。以KY131是”0/0”, 而 “DN422” 是 “1/1”的位点结果为例,我们来增加这条线
我写了一个函数用于根据窗口来计算均值,输入是之前snp-index的位置和对应的值,以及确定窗口的大小和步长,
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 calcValueByWindow <- function ( pos, value, window_size = 1000000 , step_size = 100000 ) { max_pos <- max ( pos) window_start <- seq( 0 , max_pos + window_size, step_size) window_end <- window_start + step_size mean_value <- vector( mode = "numeric" , length = length ( window_start) ) for ( j in seq_along ( window_start) ) { pos_in_window <- which( pos > window_start[ j] & pos < window_end[ j] ) value_in_window <- value[ pos_in_window] mean_value[ j] <- mean( value_in_window) } nan_pos <- is.nan ( mean_value) mean_value <- mean_value[ ! nan_pos] window_pos <- ( ( window_start + window_end) / 2 ) [ ! nan_pos] df <- data.frame( pos = window_pos, value = mean_value) return ( df) }
得到结果就可以用用lines在之前结果上加上均值线
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 par( mfrow = c ( 3 , 4 ) ) for ( i in paste0( "chr" , formatC( 1 : 12 , width = 2 , flag= 0 ) ) ) { freq_flt <- freq2[ grepl( i, row.names( freq2) ) , ] pos <- as.numeric ( substring( row.names( freq_flt) , 7 ) ) snp_index <- freq_flt[ , 1 ] - freq_flt[ , 2 ] df <- calcValueByWindow( pos = pos, value = snp_index) plot( x = pos, y = snp_index, ylim = c ( - 1 , 1 ) , pch = 20 , cex = 0.2 , xlab = i, ylab = expression ( paste( Delta, " " , "SNP index" ) ) ) lines( x = df$ pos, y = df$ value, col = "red" ) }
最后再对文章总结一下。文章并不是只用了BSA的方法进行定位,他们花了几年的时间用SSR分子标记确定了候选基因可能区间,用BSA的方法在原有基础上缩小了定位区间。当然即便如此,候选基因也有上百个,作者通过BLAST的方式,对这些基因进行了注释。尽管中间还加了一些GO富集分析的内容,说这些基因富集在某个词条里,有一个是DNA metabolic processes(GO:0006259),但我觉得如果作者用clusterProfiler做富集分析,它肯定无法得到任何富集结果。他做富集分析的目的是其实下面这个描述,也就是找到和抗冻相关的基因
LOC_Os06g39740 and LOC_Os06g39750,were annotated as the function of “response to cold (GO: 0009409)”, suggesting their key roles in regulating cold tolerance in rice. “
当然他还做了qRT-PCR进行了验证,最后推测LOC_Os06g39750应该是目标基因,这个基因里还有8个SNP位点。