这感觉就像多年前我刚开始接触RNA-seq,看书都是似懂非懂(就是那本「RNA-seq best practice」)。就跟段子写的一样,打开书,马冬梅,关上书,马什么梅?打开书,马冬梅,关上书,什么冬梅?考试,孙红雷。直到某一次生物统计课后,我在回去的路上,突然感觉一切都连接在一起,整个大脑都兴奋了起来。那一天,我才感觉自己站到了生物信息学的大门前。
另一个问题我在”Identify TIR candidates from scratch”这一步出现下面的报错
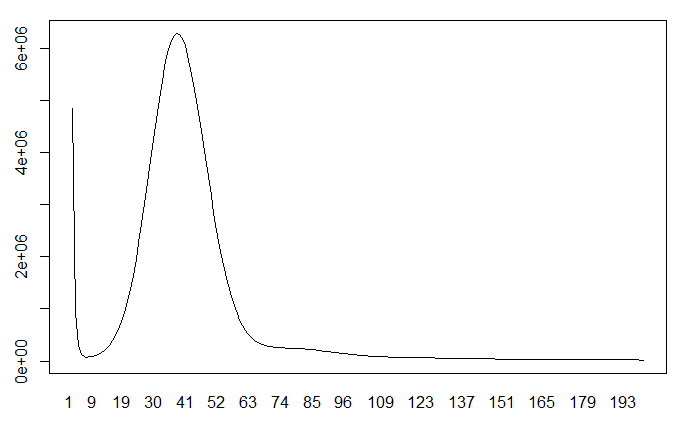
1 2 3
what(): terminate called after throwing an instance of 'Resource temporarily unavailable std::system_error' what(): Resource temporarily unavailable terminate called after throwing an instance of 'std::system_error'
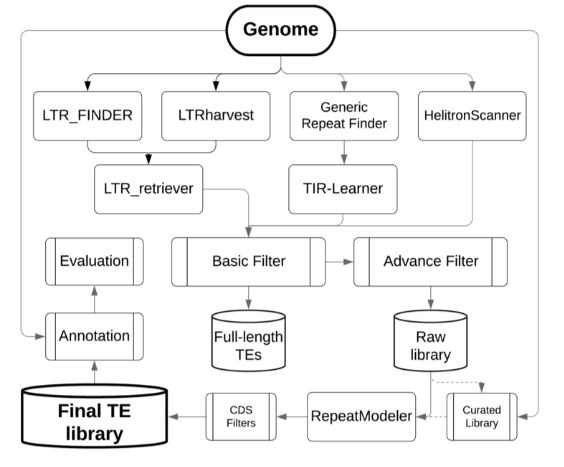
Ou, S., Su, W., Liao, Y., Chougule, K., Agda, J.R.A., Hellinga, A.J., Lugo, C.S.B., Elliott, T.A., Ware, D., Peterson, T., et al. (2019). Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biology 20, 275.
mount | grep /data2 # dev/sdb2 on /data2 type ext4 (rw,relatime,seclabel,stripe=64,data=ordered) mount -o remount,usrquota,grpquota /data2 # /dev/sdb2 on /data2 type ext4 (rw,relatime,seclabel,quota,usrquota,grpquota,stripe=64,data=ordered)
quotacheck: Your kernel probably supports journaled quota but you are not using it. Consider switching to journaled quota to avoid running quotacheck after an unclean shutdown.
如果出现下面提示,说明该磁盘正在独写,使用lsof/fuser查找可能的进程并关闭,之后重新运行。
1 2
quotacheck: Cannot remount filesystem mounted on /data2 read-only so counted values might not be right. Please stop all programs writing to filesystem or use -m flag to force checking.
下面的提示会在首次执行时,可以忽略
1 2 3 4 5 6 7 8
quotacheck: Scanning /dev/sdb1 [/data3] done quotacheck: Cannot stat old user quota file /data3/aquota.user: No such file or directory. Usage will not be subtracted. quotacheck: Cannot stat old group quota file /data3/aquota.group: No such file or directory. Usage will not be subtracted. quotacheck: Cannot stat old user quota file /data3/aquota.user: No such file or directory. Usage will not be subtracted. quotacheck: Cannot stat old group quota file /data3/aquota.group: No such file or directory. Usage will not be subtracted. quotacheck: Checked 44 directories and 23 files quotacheck: Old file not found. quotacheck: Old file not found.
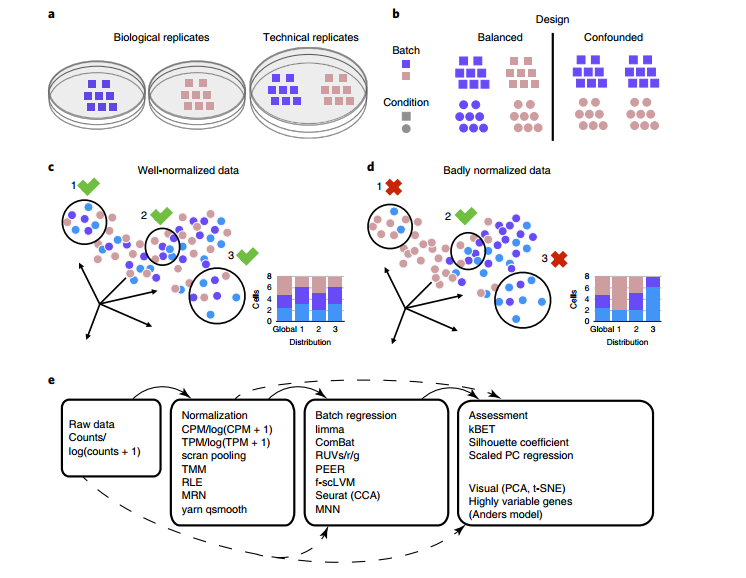
从直觉上讲,最好是分析的过程中过滤掉这种批次效应。但是即便是同一个人对同一个样本做的相同实验,也有可能因为时间差异导致批次效应,我们需要对这种数据集进行批次效应校正吗?我们对批次效应进行校正的同时也会引入新的问题,它很有可能将生物学本身的差异视为批次效应,然后将其去除。因此解决同一种实验的批次效应最好的方法就是搞一套比较好的实验设计(例如将样本分开在不同的实验批次中,(Kang et al., 2018))。但是受限于实验条件,基本上做不到这些,而且这类批次效应可能也没有那么大的影响。
Haghverdi, L., Lun, A.T.L., Morgan, M.D., and Marioni, J.C. (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 36, 421–427.
Büttner, M., Miao, Z., Wolf, F.A., Teichmann, S.A., and Theis, F.J. (2019). A test metric for assessing single-cell RNA-seq batch correction. Nat Methods 16, 43–49.
Kang, H.M., Subramaniam, M., Targ, S., Nguyen, M., Maliskova, L., McCarthy, E., Wan, E., Wong, S., Byrnes, L., Lanata, C.M., et al. (2018). Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36, 89–94.