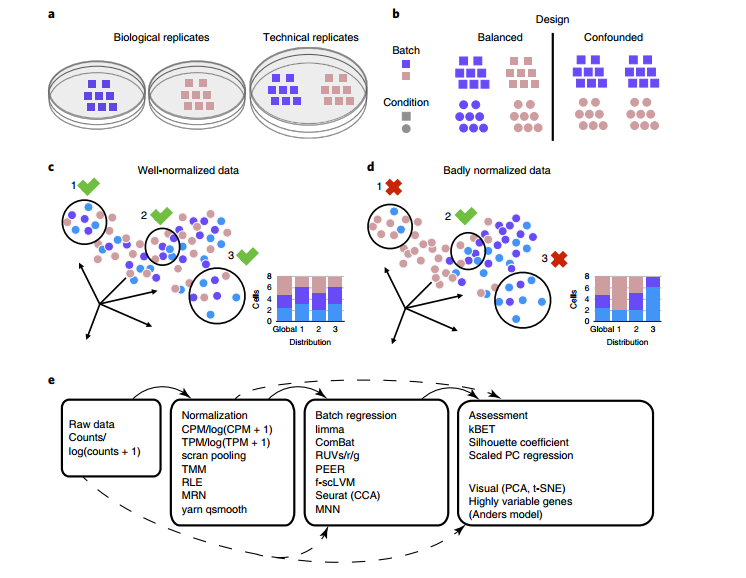
在数据分析的时候,我们的目标是找到样本之间真实的生物学差异。但是这种真实的生物学因素往往会受到各种因素影响,举几个场景
- 不同样本
- 同一样本的生物学重复
- 同一样本的技术重复
- 同一样本在同一个实验室由同一团队在不同时间点处理
- 同一细胞系/小鼠在不同实验室
- 不同建库策略,10X平台,Drop-seq, SMART2-seq
- 不同测序平台,BGI/Illumina
- 不同分析流程(甚至一个工具的多个版本,如salmon,CellRanger)
这些因素之间有些是生物学真实的差异,有些是抽样时的随机波动。有些是系统性因素,比如说批次效应(batch effect)。
从直觉上讲,最好是分析的过程中过滤掉这种批次效应。但是即便是同一个人对同一个样本做的相同实验,也有可能因为时间差异导致批次效应,我们需要对这种数据集进行批次效应校正吗?我们对批次效应进行校正的同时也会引入新的问题,它很有可能将生物学本身的差异视为批次效应,然后将其去除。因此解决同一种实验的批次效应最好的方法就是搞一套比较好的实验设计(例如将样本分开在不同的实验批次中,(Kang et al., 2018))。但是受限于实验条件,基本上做不到这些,而且这类批次效应可能也没有那么大的影响。
我们更关注的一类批次效应,其实是不同实验室,不同建库手段,不同测序平台所引起的批次效应。当我们希望通过合并同一组织数据挖掘出更有意义的信息时,就不可避免的会发现,明明是同个组织的数据,表达量就是存在明显的差异(PCA, t-SNE降维可视化)。
之前有人用bulk RNA-seq的方法(limma, ComBat,RUVseq, svaseq)对单细胞数据进行校正,但是这些工具的基本假设都是”bulk RNA-seq数据中的细胞组成相似”,可能适用于一些数据集,但是可推广性不强(Haghverdi et al., 2018)。于是就有一些专门用于单细胞转录组批次校正的工具,这里对这些软件做一个罗列
最后对于不同的实验设计,仅从主观上推荐的批次矫正方法
- 技术重复: ComBat
- 细胞系,生物学重复: ComBat
- 同一个人的癌症和癌旁组织: 不校正/Harmony
- 不同实验室的同一组织: Harmony
- 同一个实验室做的不同人的样本: 不校正/Harmony
Harmony就是给soft k-mean(GMM)加了一个KL divergence,然后这个惩罚项最小化可以使得不同batch的cells尽可能分配到每个cluster内部,然后可以给每个cell分配一个到cluster的probability,那么每个cluster都会有一个和细胞数维度一样的细胞分配到这个cluster的probability,其实就是后续一个(batch number+1)维度的向量到embedding出来向量的一个回归方程的样本权(权重线性回归),简单而言它认为将不同batch的cell embedding的向量是由batch的variation和cell type的variation所决定,只要把cell type这部分找到(其实就是残差)找到就可以去除batch的影响。但是要定义样本在每个cluster里面的entropy,而且要使这个值尽可能大,所以我不知道这个文章有没有做rare celltype相关的一些测试
-@汪伟旭(复旦大学)
对于批次效应的检查,可以试试kBET,但是它的内存和运算时间和数据集大小密切相关,很有可能跑不下去。
参考文献
- Haghverdi, L., Lun, A.T.L., Morgan, M.D., and Marioni, J.C. (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 36, 421–427.
- Büttner, M., Miao, Z., Wolf, F.A., Teichmann, S.A., and Theis, F.J. (2019). A test metric for assessing single-cell RNA-seq batch correction. Nat Methods 16, 43–49.
- Kang, H.M., Subramaniam, M., Targ, S., Nguyen, M., Maliskova, L., McCarthy, E., Wan, E., Wong, S., Byrnes, L., Lanata, C.M., et al. (2018). Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36, 89–94.