在组装基因组之前一定要先对要组装的物种有一个大致的了解,判断其复杂程度, 标准如下
- 基因组大小:基因组越大,测序花的钱越多
- 简单基因组: 杂合度低于0.5%, GC含量在35%~65%, 重复序列低于50%
- 二倍体普通基因组: 杂合度在0.5%~1.2%中间,重复序列低于50%。或杂合度低于0.5%,重复序列低于65%
- 高复杂基因组: 杂合度>1.2% 或 重复率大于65%
k-mers估计法
最简单的策略就是基于k-mer对基因组做一个简单的了解, 使用jellyfish统计k-mers,然后作图
1 | jellyfish count -m 21 -s 20G -t 20 -o 21mer_out -C <(zcat test_1.fq.gz) <(zcat test_2.fq.gz) |
一些注意事项:
- 绝对不要用
--min-qual-char或其他参数,它们会将低质量的碱基替换成N - 在测序时由于不知道测得到底是DNA的哪一条链,因此k-mer及其互补链其实是等价的,所以一定要用
-C参数
将数据导入R语言中,进行作图
1 | pdf("21_mer.out.pdf") |
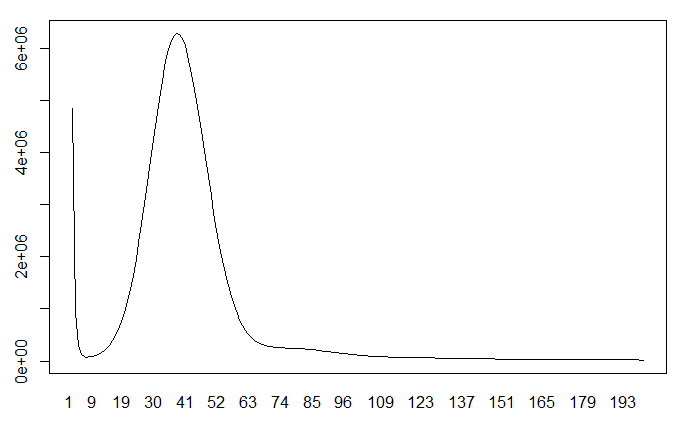
由于只有一个主峰,说明该物种的杂合度并不高,基本上也就是二倍体。如果图中出现多个峰,说明它可能是多倍体或者是基因组杂合度高。
基因组大小(G)估计算法为:
$$
G= K_{num} / K_{depth}
$$
其中 $K_{depth}$ 为K-mer的期望测序深度, $K_{num}$ 为K-mer的总数。 通常将K-mer深度分布曲线的峰值作为其期望深度。
基因组的杂合性和使得来自杂合片段的K-mer深度较纯合区段降低50%。如果目标基因组有一定的杂合性,会在k-mer深度分布曲线主峰位置(c)的1/2处(c/2)出现一个小峰。杂合度越高,该峰越明显。
推荐文献: Genomic DNA k-mer spectra: models and modalities
基于组装
基于K-mers可以较好的预测基因组大小,并定性的了解基因组的复杂情况,如果想更具体的了解基因组的复杂度,可以先将50X以上的段片段进行组装,然后进行分析。
组装的工具比较多,推荐用SOAPdenovo,因为速度快。
新建一个contig.config, 增加如下内容
1 | max_rd_len=150 |
组装出参考序列
1 | ~/opt/biosoft/SOAPdenovo2/SOAPdenovo-63mer all -s contig.config -R -K 63 -p 30 -o assembly/graph |
最后graph.scafSeq是拼接后的序列, 提取出大于300bp的序列.
1 | # adjust format |
杂合度估计
将原来的序列回贴到contig上,并用samtools+bcftools进行snp calling.统计变异的碱基占总体的比例。
1 | mkdir -p index |
一方面由于SOAPdenovo组装过程中会出错, 另一方面samtools在变异检测上也存在很高的假阳性, 所以总得先按照深度和质量过滤一批假阳性。
1 | bcftools view -i ' DP > 30 && MQ > 30' -H variants.vcf.gz | wc -l |
变异数目占基因组大小的比例就是杂合度。我的contig大概是200M,找到0.3M左右的变异,也就是0.0015,即0.15%.
重复序列估计
基于同源注释,用RepeatMasker寻找重复序列. 这里要注意分析的fasta的ID不能过长,也就是最好是>scaffold_1这种形式,不然会报错。
1 | ~/opt/biosoft/RepeatMsker/RepeatMasker -e ncbi -species arabidopsis -pa 10 -gff -dir ./ contig.fa |
输出结果中主要关注如下三个
- output.fa.masked, 将重复序列用N代替
- output.fa.out.gff, 以gff2形式存放重复序列出现的位置
- output.fa.tbl, 该文件记录着分类信息
1 | ================================================== |
也就是说我们的物种有30%的重复序列,作为参考,拟南芥125Mb 14%重复序列, 水稻389M,36%重复
附录:软件安装
安装RepeatMasker
1 | cd ~/src |