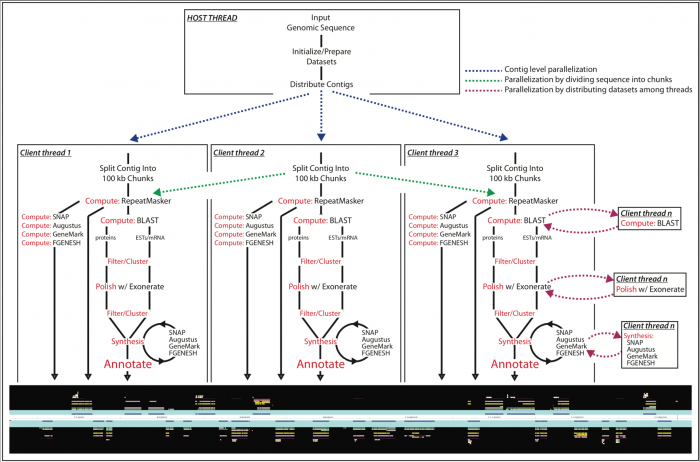
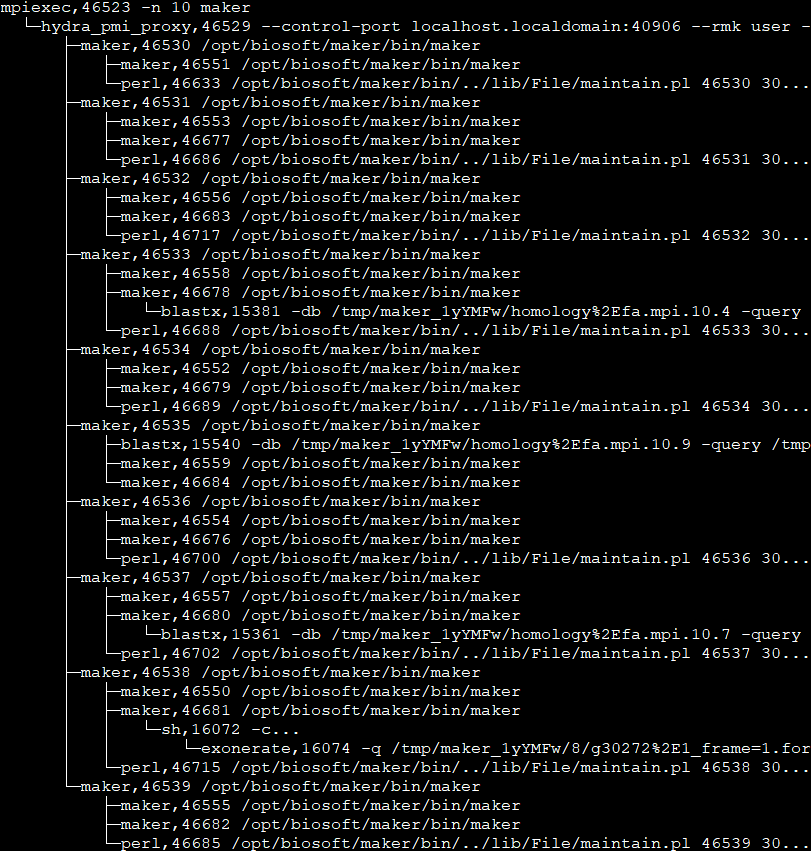
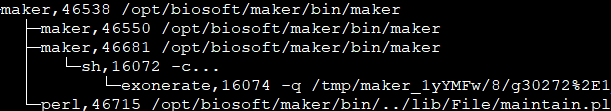
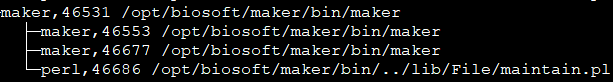
从中你可以发现,因为我们用mpiexec启动了10个maker进程,所以hydra_pmi_proxy有10个maker子进程。对于这10个maker子进程,每个进程都至少会有一个maintain.pl。通过阅读maintain.pl的源代码,我们可以得知该程序后接参数中的46535是PID(进程号), 30是sleep time, 最后一个是URI编码字符,是利用Storable::freeze持久化的数据结构,可以通过如下的代码进行解码(命名为decode.pl)
1 2 3 4 5 6 7 8 9 10 11 12 13 14
#!/usr/bin/env perl use warnings; use strict;
use URI::Escape; use Storable; use vars qw($LOCK);
# Information about the version of MPICH to use export MPICH_VERSION=3.3 export MPICH_URL="http://www.mpich.org/static/downloads/$MPICH_VERSION/mpich-$MPICH_VERSION.tar.gz" export MPICH_DIR=/opt/mpich
echo"Installing MPICH..." mkdir -p /tmp/mpich mkdir -p /opt # Download cd /tmp/mpich && wget -O mpich-$MPICH_VERSION.tar.gz $MPICH_URL && tar xzf mpich-$MPICH_VERSION.tar.gz # Compile and install cd /tmp/mpich/mpich-$MPICH_VERSION && ./configure --prefix=$MPICH_DIR && make install # Set env variables so we can compile our application export PATH=$MPICH_DIR/bin:$PATH export LD_LIBRARY_PATH=$MPICH_DIR/lib:$LD_LIBRARY_PATH export MANPATH=$MPICH_DIR/share/man:$MANPATH
echo"Compiling the MPI application..." cd /opt && mpicc -o mpitest mpitest.c
cM <- confusionMatrix(paste0(projHeme2$Clusters), paste0(projHeme2$Sample)) cM
文字信息太多,这里直接用热图进行展示
1 2 3 4 5 6 7 8
library(pheatmap) cM <- cM / Matrix::rowSums(cM) p <- pheatmap::pheatmap( mat = as.matrix(cM), color = paletteContinuous("whiteBlue"), border_color ="black" ) p
projHeme1 <- ArchRProject( ArrowFiles = ArrowFiles, outputDirectory ="HemeTutorial", copyArrows =TRUE#This is recommened so that if you modify the Arrow files you have an original copy for later usage. )
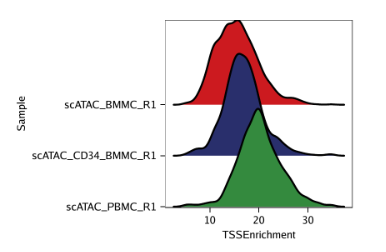
p1 <- plotGroups( ArchRProj = projHeme1, groupBy ="Sample", colorBy ="cellColData", name ="TSSEnrichment", plotAs ="ridges" ) p1
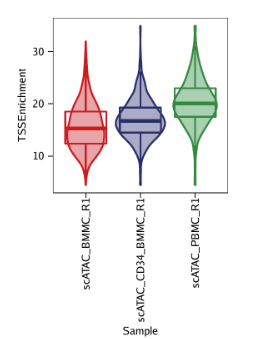
例2: 根据TSS富集得分为每个样本绘制小提琴图。ArchR绘制的小提琴图额外叠加了一层箱线图(a box-and-whiskers plot in the style of Tukey),中间水平的三条线表示数据中的1/4分位数,中位数和3/4分位数。最下方是最小值,最上方是最大值(或者是1.5倍的IQR, interquartile range, 1/4分位数与3/4分位数的距离)
绘制小提琴图时,需要设置plotAs = "violin"
1 2 3 4 5 6 7 8 9
p2 <- plotGroups( ArchRProj = projHeme1, groupBy ="Sample", colorBy ="cellColData", name ="TSSEnrichment", plotAs ="violin", alpha =0.4, addBoxPlot =TRUE )
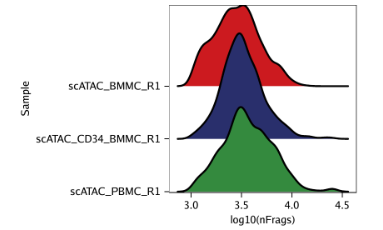
例3: 根据log10(unique nuclear fragments)为每个样本绘制山脊图。
1 2 3 4 5 6 7 8
p3 <- plotGroups( ArchRProj = projHeme1, groupBy ="Sample", colorBy ="cellColData", name ="log10(nFrags)", plotAs ="ridges" ) p3
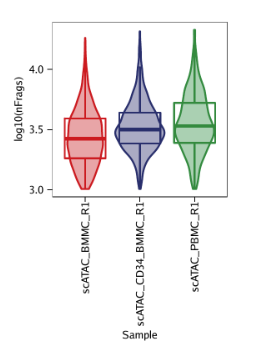
例4: 根据log10(unique nuclear fragments)为每个样本绘制小提琴图。
1 2 3 4 5 6 7 8 9 10
p4 <- plotGroups( ArchRProj = projHeme1, groupBy ="Sample", colorBy ="cellColData", name ="log10(nFrags)", plotAs ="violin", alpha =0.4, addBoxPlot =TRUE ) p4
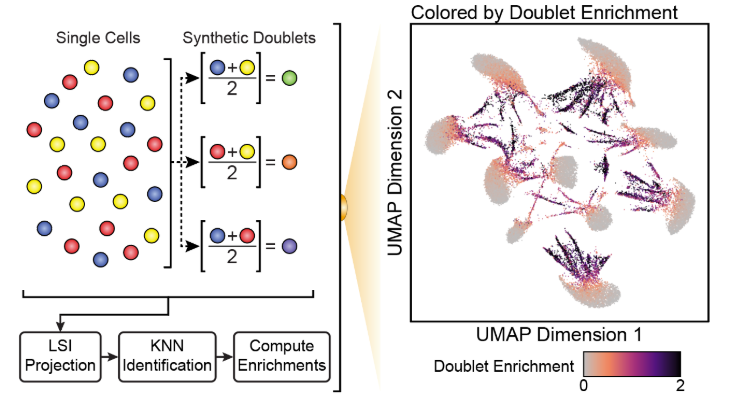
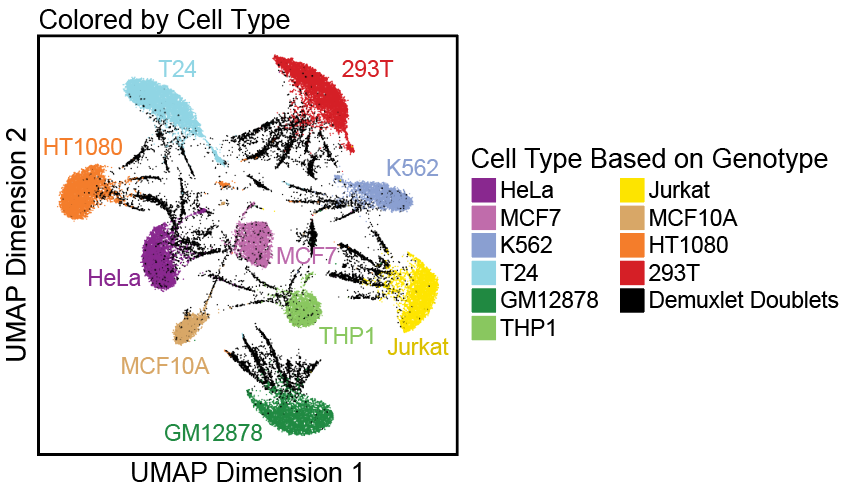
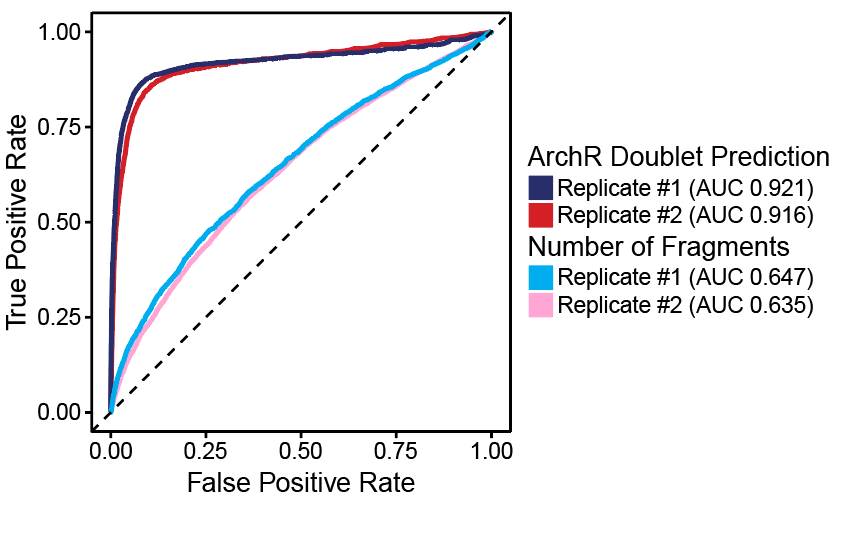
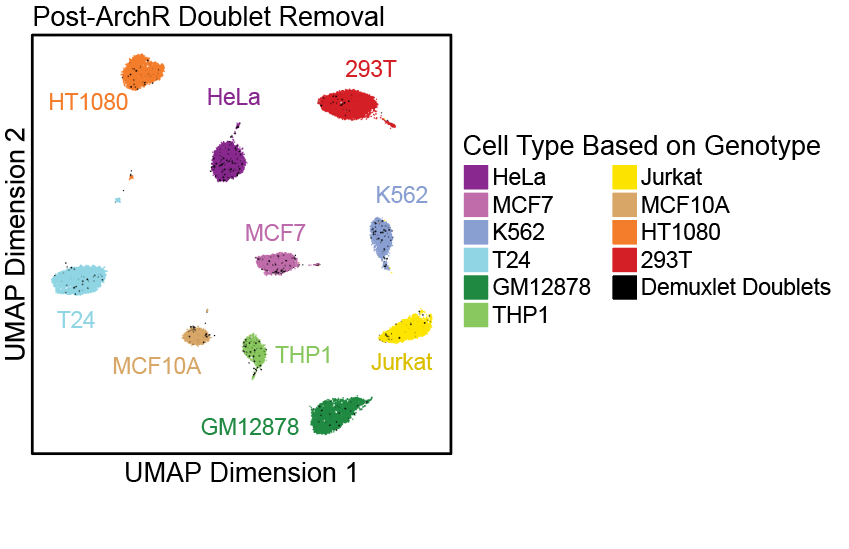
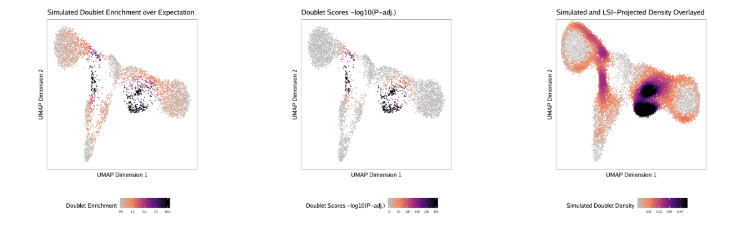
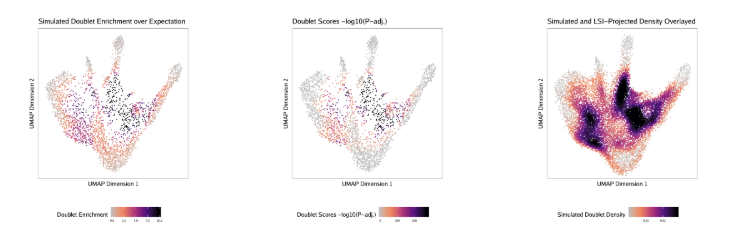
了解了filterRatio的作用后,我们再来看它的定义,它是根据未被过滤的细胞数计算的需要被过滤doublet的最大值(the maximum ratio of predicted doublets to filter based on the number of pass-filter cells)。举个例子。假如这里有5000个细胞,需要被过滤doublet的最大值就等于 filterRatio * 5000^2 / (100000), 也就是filterRatio * 5000 * 0.05。
doubScores <- addDoubletScores( input = ArrowFiles, k =10,#Refers to how many cells near a "pseudo-doublet" to count. knnMethod ="UMAP",#Refers to the embedding to use for nearest neighbor search with doublet projection. LSIMethod =1 )