第4章: ArchR降维分析
scATAC-seq数据的稀疏性让降维这一步充满了挑战性。在scATAC-seq数据中,一个特定的位置存在三种状态,分为不开放,一条链开放,两条链都开放。但即便是在高质量的scATAC-seq数据中,大部分开放区域都没有被转座酶切割,因此大部分座位都是0。此外,如果我们发现一个细胞(A)的一处peak区域有三个Tn5 insertions,而另外一个细胞(B)对应区域只有一个Tn5 insertions,由于数据的稀疏性,我们也无法断定A就比B更开放。鉴于此,许多分析框架都会把scATAC-seq数据矩阵二值化,也就是只有0和1两种情况。即便如此,因为转座酶切割的位置还是很少,所以二值矩阵大部分区域还是0。我们还需要注意的是,scATAC-seq数据中的0既有可能表示”不开放”,也可能是”没取样到”, 从生物学角度上出发,这是两种截然相反的推断。正因为数据大部分都是0,所以1比0更有信息量。scATAC-seq数据的稀疏性便是来源于这种低信息量。
如果你直接在稀疏矩阵上使用标准的降维分析,例如主成分分析(Principal Component Analysis, PCA),然后使用前两个主成分进行绘图,你可能无法得到你想要的结果,这是由于数据的稀疏性会导致细胞间在0的位置有更高的相似性。为了解决这个问题,我们采用了分层的降维策略。
首先,我们会使用隐语义分析(Latent Semantic Indexing, LSI)进行降维。LSI方法最早用在自然语言分析中,根据字数来评估文档相似度。之所以在自然语言分析中发明该方法,是因为文档集中会有许多不同的词,每个词出现的次数也很少,最终的数据非常稀疏且充满噪音。Cusanovich et al. (Science 2015)第一次在scATAC-sq数分析中引入了LSI。在scATAC-seq场景下,不同样本就是不同的文档,不同的区域/peak就是不同的单词(注1)。计算步骤如下
- 对每个细胞计算根据深度标准化后的词频(Term frequency, TF)
- 根据逆文档频率(IDF, inverse document frequency)进行标准化,便于后续特征选择
- 最后对结果矩阵进行对数变换(也就是
log(TF-IDF))
接着,奇异值分解( singular value decomposition, SVD)技术能够分析出不同样本间更有价值的信息,从而降低了维度。上面这两步能够将原本成千上万维的稀疏矩阵的降维到数十个或者数百个维度。
最后,我们使用更加常见降维方法,例如UMAP( Uniform Manifold Approximation and Projection ) 或t-SNE( t-distributed stochastic neighbor embedding ) 对数据进行可视化。ArchR称这这些可视化方法为嵌入(embeddings, 注2)
- 注1: 对LSI最直观理解就是它在自然语言分析的用途,也就是为每篇文档找到关键词,这个关键词必须要在当前文档中出现,同时最好别在其他文档里出现。在scATAC-seq的语境中,如果一个区域在所有细胞中都有insertion,那么就不特异,最好是只有几个细胞有insertion,而大部分细胞没有,这才算一个特异的insertion。
- 注2: 我们可以认为道路是嵌入在三维空间中一维流形。我们用一维道路中的地址号码确定地址,而非三维空间的坐标。
4.1 ArchR的LSI实现
ArchR实现了多种LSI方法,并用不同测数据集对这些方法进行了测试。ArchR默认的LSI实现和Timothy Stuart在Signac引入的方法有关,也就是先将词频(TF, term frequency)根据常数(10,000)进行深度标准化,然后根据逆文档频率(IDF, inverse document frequency)进行标准化,最后对结果矩阵进行对数变换(也就是log(TF-IDF))
LSI降维的其中一个关键输入是起始矩阵。到目前为止,scATAC-seq主要有两种策略计算矩阵,一是使用peak区域,二是对全基因组分块(genome-wide tiles)。由于在降维前,我们还没有聚类,也没有聚类特异的peak,所以不能使用peak区域作为LSI的输入。而且直接使用所有数据进行peak calling,会导致一些细胞特异性的peak被掩盖。更何况,当你在实验中增加新的样本,那么合并后的peak集就会发生改变,导致整个方法不太稳定。第二种策略通过对全基因组分块,保证了特征集(feature set)的一致性和无偏好性,缓解了第一种方法存在的问题。只不过,使用全基因组分块会得到一个非常大的细胞X特征的矩阵,因此大部分LSI实现都至少用5000 bp对基因组进行分块。计算量降低的同时也导致分辨率急剧下降,因为大部分开放区域只有几百个碱基的长度。
在Arrow文件的设计帮助下,即便是以500-bp作为基因组的分块,ArchR依旧能够快速的计算LSI。这就解决了分辨率的问题,使得在calling peak前就能进行聚类分析。使用500-bp进行分块的更大挑战是这会得到一个拥有数百万个特征的cell-by-tile矩阵。尽管ArchR能够通过读取必要矩阵的方式在R中加载部分数据,但是我们还是实现了一个”近似LSI”算法,使用一部分细胞进行最初的降维分析。近似LSI有两个主要用途,(1)提高降维速度,(2)初始降维时使用的细胞数的减少会降低数据的粒度,而这种粒度的减少有利于减少数据中的批次效应。但要注意,这可能会掩盖真实的生物学现象,因此近似LSI方法需要在人为监督下谨慎的使用。
4.2 隐语义(Latent Semantic Indexing)迭代
在scRNA-seq降维分析中(例如PCA),一般都会鉴定变异基因。之所以这样做,是因为高变异基因通常更有生物学意义,并能够减少实验噪音。scATAC-seq数据只有0/1两种情况,所以你不能为降维分析提供变异peak。除了无法使用变异更大的peak,我们也尝试使用了更加开放的特征作为LSI的输入,然而经过多个样本的测试,我们发现处理结果存在较高的噪音,并且很难重复。为了解决该问题,我们提出了”迭代LSI法” (Satpathy*, Granja* et al. Nature Biotechnology 2019和 Granja*, Klemm* and McGinnis* et al. Nature Biotechnology 2019). 这个方法首先在最开放的分块中计算初始的LSI变换,识别未受到批次效应影响的低分辨率聚类。例如,我们可以在PBMC中鉴定出几个主要大群(T细胞、B细胞和单核细胞)。接着,ArchR计算这些聚类在每个特征中的平均开放度。ArchR识别这些聚类中变化最大的peak,用作后续LSI的特征。在第二次迭代中,变化最大的peak就类似于scRAN-seq中高变异基因。用户可以决定进行多次轮的LSI迭代。我们发现这个方法能够降低观察到的批次效应,并能使用更加合理的特征矩阵进行降维。
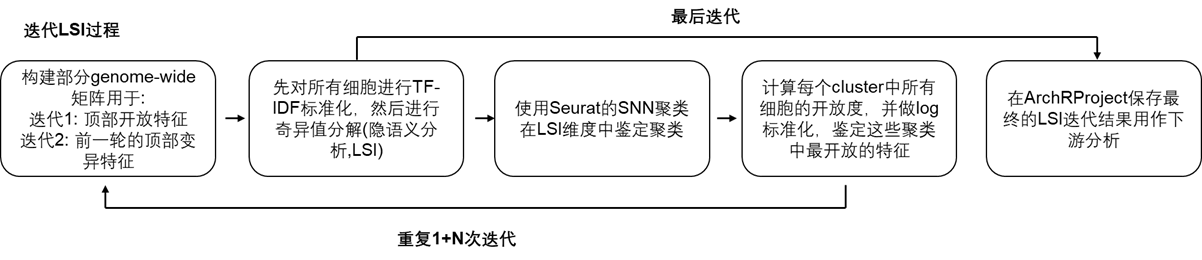
我们可以是用ArchR的addIterativeLSI()函数执行迭代LSI分析。虽然默认参数适用于绝大部分情况,但是我们还是鼓励你使用?addIterativeLSI看看有哪些可用的参数,以及它们对你的数据集的影响。一般经常修改的参数是iterations,varFeatures和resolution。注意,LSI的结果并不是确定的,也就是即便你用相同参数处理同一个数据,也会得到不一样的结果。当然只是略微不同,总体上还是相似的。因此,一旦你得到一个相对理想的降维结果,请及时保存你的ArchRProject项目或者相关的LSI信息。
处于教学目的,我们会创建一个名为”IterativeLSI”的reduceDims对象
1 | projHeme2 <- addIterativeLSI( |
如果你在下游分析时看到一些批次效应,其中一个选择就是增加LSI的迭代次数,并以更低的分辨率进行聚类。此外,也可以通过降低特征变量的数目让程序关注变异更大的特征。
1 | projHeme2 <- addIterativeLSI( |
4.3 近似LSI
对于特别大的数据集,ArchR使用LSI投影得到近似的LSI降维结果。这个步骤和之前的迭代LSI流程相似,但是在LSI步骤存在一些不同。首先,我们会随机选择一部分”路标细胞”用于LSI降维分析。接着,使用”路标细胞”计算的逆文档频率对余下的细胞进行TF-IDF标准化。然后,这些标准化的细胞被投影到”路标细胞”定义的SVD子空间中。最终,我们就将余下的细胞根据少量的”路标细胞”实现了LSI变换和投影。因为ArchR不需要将所有细胞的read都保存在内存中,而是不断的提取每个样本的细胞,将其投影到路标细胞的LSI空间中,所以近似LSI算法非常高效。因此,即便之后用到了更大的数据集,近似LSI也能够减少内存的使用。
注意,路标细胞的数量取决于数据集中不同细胞的比例。
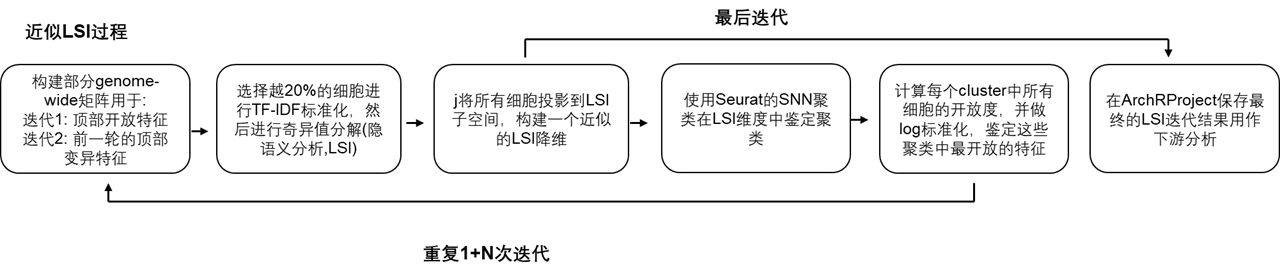
近似LSI通过修改ArchR的addIterativeLSI()函数的sampleCellsFinal和projectCellsPre参数来实现.samplesCellsFinal决定了路标细胞的数目,projectCellsPre告诉ArchR使用路标细胞对其余细胞进行投影。
4.4 使用Harmony矫正批次效应
在某些情况下,迭代的LSI方法并不能矫正严重的批次效应。ArchR使用了最初为了scRNA-seq开发的Harmony进行批次效应矫正。 ArchR提供了专门的封装函数addHarmony()将降维后的对象直接传递给Harmony::HarmonyMatrix(), 额外参数可以通过...进行传递。更详细的信息可以用?addHarmony()进行了解。用户需要根据他们的特定目的注意批次效应校正的结果。
1 | projHeme2 <- addHarmony( |
这一步会在我们的projHeme2创建一个新的名为Harmony的reduceDims对象,可以通过projHeme2@reducedDims$Harmony访问。