背景篇
在植物基因组中,I类转座因子,LTR-RT(LTR retrotransposons)是基因组扩张的主要原因。完整的LTR长度在855000 bp之间,下图图A表示的是一个完整的LTR-RT,灰色框表示TSD(target site duplications), 红色三角形表示LTR motif(长度在2bp左右), 蓝色框表示LTR。LTR中间序列长度在1,00015,000之间波动。
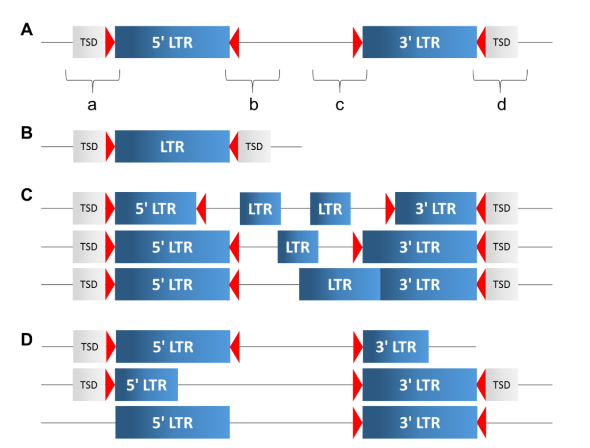
完整的LTR-RT主要归为两大类: Gypsy和Copia。如果LTR中间的序列不包含开放阅读框(ORF), 那么所属的LTR-RT就无法独立的转座。
安装篇
LTR_retriever不是一个独立的工具,他的主要作用就是整合 LTRharvest, LTR_FINDER, MGEScan 3.0.0, LTR_STRUC, 和 LtrDetector的结果,过滤其中的假阳性LTR-RT,得到高质量的LTR-RT库。
LTR_retriever托管在GitHub, https://github.com/oushujun/LTR_retriever, 下载LTR_retriever本体
1 | git clone https://github.com/oushujun/LTR_retriever.git |
之后修改LTR_retriever下的paths, 提供BLAST+, RepeatMasker, HMMER, CDHIT这些工具的路径。
1 | BLAST+=/your_path_to/BLAST+2.2.30/bin/ |
更加方便的安装方法用Bioconda安装好cd-hit repeatmasker, 然后下载LTR_retriever:
1 | conda create -n LTR_retriever |
此外你还需要额外安装LTRharvest, LTR_FINDER 和MGEScan_LTR。
- LTRharverst: http://genometools.org/
- LTR_FINDER: https://github.com/xzhub/LTR_Finder
- 修改版MGEScan_LTR: http://dawgpaws.sourceforge.net/
由于MGEScan_LTR装起来比我想象中麻烦,所以本文就仅使用LTRharverst和LTR_FINDER
使用篇
尽管LTR_retriever支持多个LTR工具的输入,但其实上LTRharverst和LTR_FINDER的结果就已经很不错了。
以拟南芥的基因组序列为例,分别使用LTRharverst和LTR_FINDER来寻找拟南芥中潜在LTR序列,之后用LTR_retreiver来合并结果。
1 | #LTRharvest |
LTR_retriever支持单个候选的LTR,
1 | LTR_retriever -genome TAIR10.fa -inharvest TAIR10.harvest.scn |
也支持多个候选LTR输入
1 | LTR_retriever -genome TAIR10.fa -inharvest TAIR10.harvest.scn -infinder TAIR10.finder.scn -threads 20 |
输出文件如下
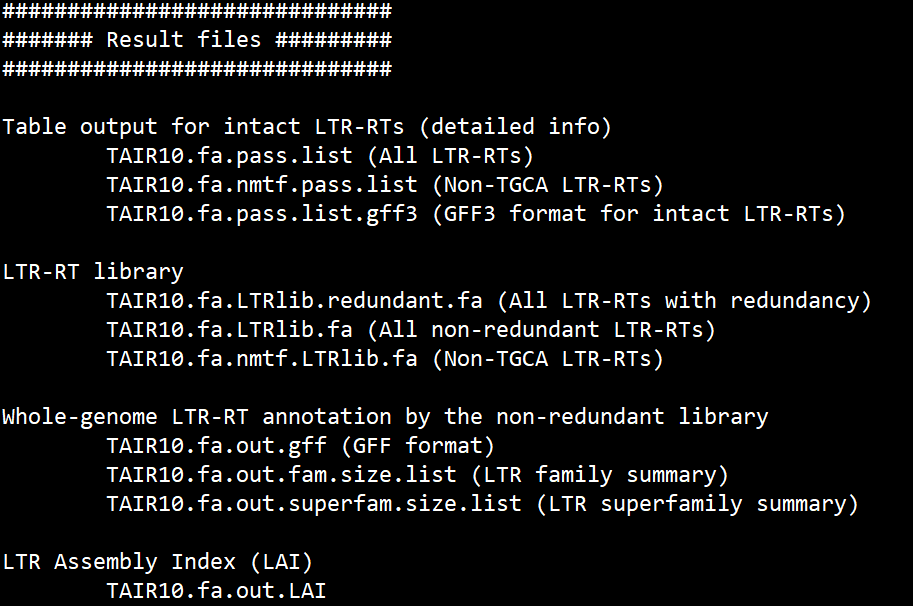
其他测试
LAI值是作者提出用于衡量基因组完整度参数。比较2个LTR输入和1个LTR输入的LAI值,后者是15.62,前者是14.47,这也意味这个值其实是受到输入的候选LTR数目影响,但最终结果应该稳定在一个阈值内。
我测试了多个物种在两种软件下找到的LTR,以及最终pass留下的LTR, 发现最终能够pass,数量都相对较少。同时限速步骤就是LTR_finder 和 LTRharvest。
| 物种 | 基因组大小 | LTR_finder | LTRharvest | Pass | LAI | 测序技术 |
|---|---|---|---|---|---|---|
| A. lyrata | 206M | 1456 | 1017 | 1044 | 20.39 | Sanger |
| A. thaliana (TAIR10) | 120 M | 207 | 550 | 184 | 15.62 | Sanger |
| B. rapa (2.5) | 391M | 1251 | 3182 | 520 | 0 | PacBio + 二代20Kb 40Kb文库 |
| B. rapa (3.0) | 353 M | 3515 | 3635 | 1968 | 7.16 | PacBio + BioNano + Hi-C |
| C.rubella | 135 M | 643 | 600 | 144 | 10.96 | 454 + Sanger |
| A. alpina | 336 M | 3840 | 3107 | 2556 | 11.01 | PacBio + BioNano + Hi-C |
| 某物种A | 454 M | 5384 | 2789 | 4294 | 17.89 | PacBio |
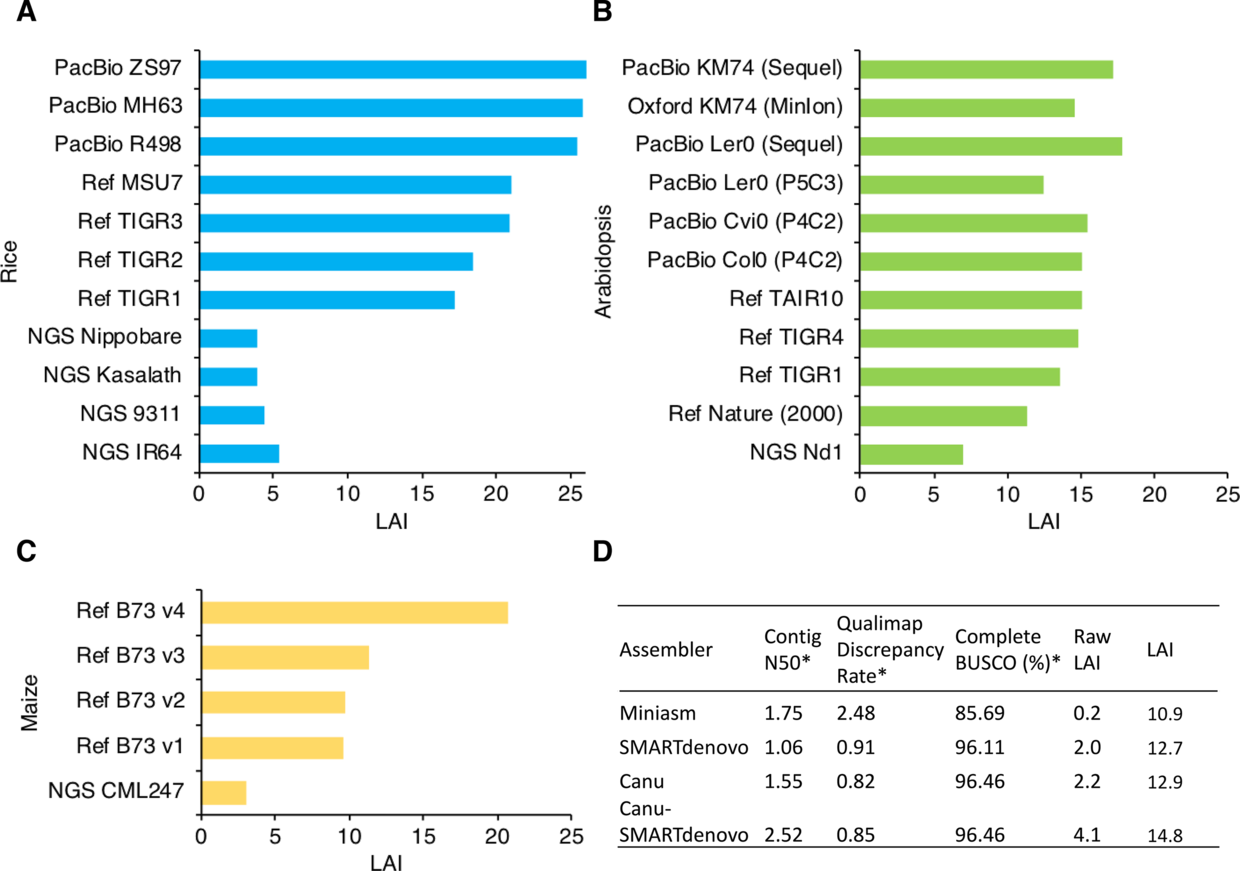
还有一个有趣的现象,B. rapa 3.0版本尽管是最近用三代加Hi-C组装的基因,但是以LAI的标准,只能算是Draft级别, 当然也比2.5版本好出不少。
当然作者也对很多物种的多个版本组装进行了比较,下图来自于 Assessing genome assembly quality using the LTR Assembly Index (LAI)
如果使用该软件记得引用下面两篇文献
- LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons
- Assessing genome assembly quality using the LTR Assembly Index (LAI)