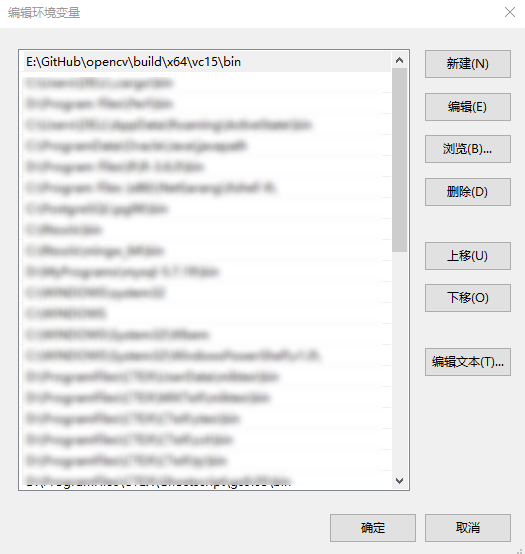
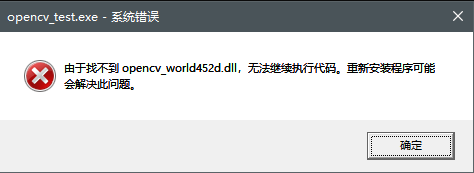
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
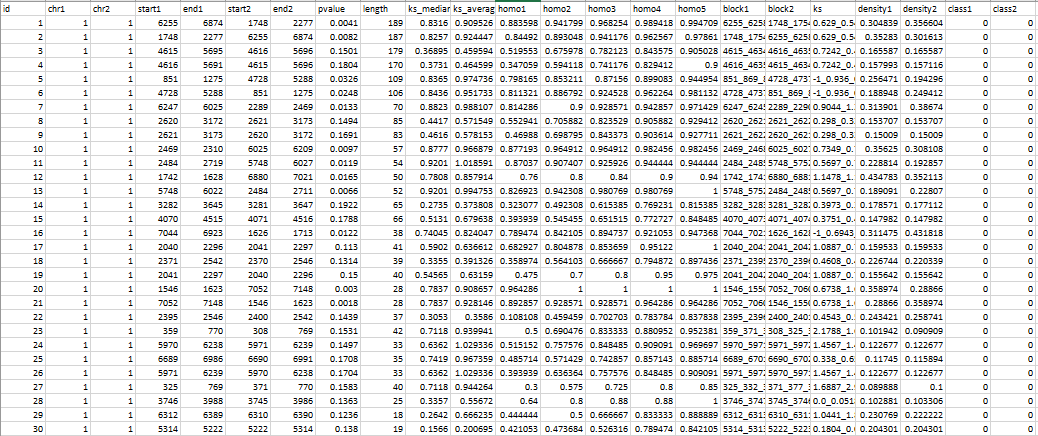
| def block_position(self, collinearity, blast, gff1, gff2, ks):
data = []
for block in collinearity:
blk_homo, blk_ks = [], []
if block[1][0][0] not in gff1.index or block[1][0][2] not in gff2.index:
continue
chr1, chr2 = gff1.loc[block[1][0][0],
'chr'], gff2.loc[block[1][0][2], 'chr']
array1, array2 = [float(i[1]) for i in block[1]], [
float(i[3]) for i in block[1]]
start1, end1 = array1[0], array1[-1]
start2, end2 = array2[0], array2[-1]
block1, block2 = [], []
for k in block[1]:
block1.append(int(float(k[1])))
block2.append(int(float(k[3])))
if k[0]+","+k[2] in ks.index:
pair_ks = ks[str(k[0])+","+str(k[2])]
blk_ks.append(pair_ks)
elif k[2]+","+k[0] in ks.index:
pair_ks = ks[str(k[2])+","+str(k[0])]
blk_ks.append(pair_ks)
else:
blk_ks.append(-1)
if k[0]+","+k[2] not in blast.index:
continue
blk_homo.append(
blast.loc[k[0]+","+k[2], ['homo'+str(i) for i in range(1, 6)]].values.tolist())
ks_arr = [k for k in blk_ks if k >= 0]
if len(ks_arr) == 0:
ks_median = -1
ks_average = -1
else:
arr_ks = [k for k in blk_ks if k >= 0]
ks_median = base.get_median(arr_ks)
ks_average = sum(arr_ks)/len(arr_ks)
df = pd.DataFrame(blk_homo)
homo = df.mean().values
if len(homo) == 0:
homo = [-1, -1, -1, -1, -1]
blkks = '_'.join([str(k) for k in blk_ks])
block1 = '_'.join([str(k) for k in block1])
block2 = '_'.join([str(k) for k in block2])
data.append([block[0], chr1, chr2, start1, end1, start2, end2, block[2], len(
block[1]), ks_median, ks_average, homo[0], homo[1], homo[2], homo[3], homo[4], block1, block2, blkks])
data = pd.DataFrame(data, columns=['id', 'chr1', 'chr2', 'start1', 'end1', 'start2', 'end2',
'pvalue', 'length', 'ks_median', 'ks_average', 'homo1', 'homo2', 'homo3',
'homo4', 'homo5', 'block1', 'block2', 'ks'])
data['density1'] = data['length'] / \
((data['end1']-data['start1']).abs()+1)
data['density2'] = data['length'] / \
((data['end2']-data['start2']).abs()+1)
return data
|