我的Rust第一课
在开始之前,先得做一些基础配置。
安装Rust工具链
1 | curl --proto '=https' --tlsv1.2 -sSf https://sh.rustup.rs | sh |
我使用VScode进行开发,并安装了如下插件
- rust-analyzer
- rust syntax
- crates
- better toml
- rust test lens
- Tabnine
接着,尝试使用Rust构建第一个程序。我的目标是读取Fasta,统计其中的A, T, C, G各有多少个。
完成这个需求,有两个思路,第一个是调用成熟的库,第二个是自己造轮子写一个函数。
对于思路1,我使用关键词,Rust和Bio搜索到一个库叫做,rust-bio。 https://rust-bio.github.io/
接下来,使用cargo new创建项目
1 | cargo new fasta_count |
第二步: 添加依赖。 编辑 Cargo.toml , 添加依赖信息
1 | [dependencies] |
第三步: 学习rust-bio文档,查找相关函数,然后在 src/main.rs中编写代码。
1 | use bio::io::fasta; |
我使用标准库io::stdin从标准输入中读取数据,然后利用fasta::Reader的new出一个新的reader实例。对该实例的records对象进行遍历,得到fasta的中记录,record。最后对记录中序列进行遍历,得到不同碱基的数目。
第四步:编译代码。cargo会先将依赖环境下载到当前项目下,然后在进行编译。
1 | cargo build |
编译结果存放在 target的debug目录下,名为fasta_count。我们运行程序,得到结果。
1 | cat TAIR10.fa | target/debug/fasta_count |
第一种思路的代码,是我看了极客时间「陈天·Rust编程第一课」中第一个示例”使用HTTP获取HTML然后保存成Markdown”后自己尝试的第一个和生信相关程序。对于第二种思路,我个人还是决定后续学习更多Rust基础再来尝试,因为我目前都不会用基础库来读取文件。
目前我的初体验是,Rust的语法更接近C/C++,它的编译器非常强大,能够很好的指出的我代码中的错误,便于检索debug。
谈谈Markdown语法中段内换行
在使用markdown语法写作时,我们会发现Markdown编辑器的展示效果和网页的展示效果并不一致。
在markdown中,我们会用一个空行表示一个段落的结束。不同段落之前会换行。
如果没有空行,那么即便使用回车键换行,仍旧是一段,不会换行。
上面这段话在编辑中被分成了两行(如下图),但是在网页展示中却应该是一段,前后相连。

但此时就有会有人提出一个需求说,我能不能让段落里面有三句话,这三句话每一句话都是另起一行,但属于一个段落。
这个貌似合理的需求,默认所有人用的都是相同的浏览器,所以所有人看到的都是3行,一行一句话。但实际上,如果你把浏览器放大几倍,那么原来的一行就会因为空间不够而自动换行。
之所以在编辑中允许段落换行,是因为某些编辑器不支持自动换行。也就是如果你不人为换行,那么这个一段就真的是一行。即,编辑器的段内换行是为了方便编辑。
那么如果你
一定要实现段内换行。我们就只能使用html语法了。
上面这段话的对应的代码是
1 | 那么如果你<br>一定要实现段内换行。</br>我们就只能使用html语法了。 |
也就是利用 <br> </br>这个标签,将中间的文字在段落中另起一行。我个人特别不喜欢这种做法。因为我认为既然是一个段落,他的前后语义需要前后衔接。我们看完上一句话,会自然而然的看下一句话。那么让浏览器自动根据访问设备大小自动对段落进行换行,是一个非常合理的设计。
但是既然有了这个标签,说明它也是有一定用途。我觉得它最大使用场景就是展示诗了。
毕竟
我们都是诗人
Love and peace
记录近年来我的个人博客托管的折腾之旅
最初(2017年左右)我用的是静态网页+GitHub Page的方式搭建我的个人网站,但是后来发现这种方式有点麻烦,所以就放弃了。
后来我发现了 Halo, 一个比较轻量的博客管理系统,所以我就使用了这个工具进行博客搭建。虽然博客的发布变得非常顺利,但是由于Halo是基于Java开发的,所以需要一个服务器进行托管。因此,如何托管我的博客站点就成了一个头疼的问题。
最初我用的是bandwagonhost托管我的站点,优点是特别便宜,缺点是服务器性能有点差,同时因为在国外,所以访问速度令人堪忧。后来,云伐科技感谢我对他们服务的早期推广提供了一台免费的服务器,优点是,4核心16GB内存,性能非常过关,缺点还是访问速度,我在国内访问我自己博客有些时候要加载10s以上,自己都受不了,更别说其他人了。再后来,我学会了使用NGINX,于是用了课题组的服务器托管了站点,通过反向代理的方式,使得同一个IP地址能同时托管自己的课题组网页和自己博客,于是国内访问速度上来了,简直完美。唯独没想到的是,出于所内的网络安全问题,这种方式被叫停了。再接着,我想到了花生壳,能够允许动态IP,于是我花了500多人民币(一年的域名服务,一年的商业版穿透服务),以为自己终于要解脱。然而万万没想到的是,花生壳不支持 xuzhougeng.top 形式的域名,最最搞笑的是,这还是我排除了任何可能性,才问出来的。(他们的客服人员只会三板斧,重启你的花生壳服务器,重新登录你的花生壳账号,试试改改你DNS服务器。)
因此,我只能做到让大家通过 xuzhougeng.top 来访问我的站点。同时让对于哪些先访问到 blog.xuzhougeng.top 的小伙伴,我就利用GitHub Page的免费服务,在里面写上一条,请大家异步新站点。
在这期间,我想着,要不自己买个腾讯云服务器吧,大不了就是备案,毕竟我就是发表一些技术性文章,能有啥危险的呢?于是,我就花了1000块下单了服务器(3年),然后准备备案。但是,我没想到的是在上海备案需要居住证,我哪有这证明啊。备案这条路也被封死了。(服务器已经退款了,所以只是浪费了一段时间而已)
最后,我想着国内的服务器不行,那我就找国外的虚拟主机服务吧,所以我就找到之前用来买域名的 NameCheap. 最初我是冲着买VPS去的,但是一顿操作下来,我买的却是WordPress的托管服务。只能说我太着急了,于是我就申请退款了,重新下单了虚拟主机服务服务(原价700多,第一年优惠价格500,和国内的云服务器没有优惠的价格差不多)。中间又出了点麻烦,国外的服务界面做的不如国内界面,导致我没有找到虚拟主机服务登录的账户密码,差点又以为自己犯傻了。
不过,最终,各位能看到这篇文章,就说我终于结束了这趟折腾之旅(大概?)。
最后总结一下:
- WordPress虽然复杂,但是项目成熟,托管便宜。
- 国内的备案要求的信息还是挺多的,如果怕麻烦还是用国外的虚拟主机服务
一次路由器刷固件安插件的折腾之旅
故事的起因可以追溯到好几年之前,当时机缘巧合之下和Leo@China一起拼团买了一个二手斐讯路由器(关于这个路由器,还有一个金融理财事件在里面),第一次听说路由器刷固件的事情。
最近为了改善我们课题组的上网环境(人多设备多,旧的路由器容易断连),斥巨资购买了华硕路由器,型号为RT-AX86U,在给路由器设置时,发现界面有些眼熟,不由得让我想起来之前那台斐讯路由器的后台,于是我就想着,这台路由器是不是也能刷固件呢?
经过一波上网检索,我最终折腾成功,给这台路由器刷上了固件,离线安装了个别插件,如下是具体的教程。
如果没有特别的上网需求,不建议刷固件。
第一步,到KoolShare论坛的固件下载服务器, https://firmware.koolshare.cn/, 选择游客所能下载的最新固件。我购买的是2021年的RT-AX86U, 出场的固件版本就是386,因此我在 https://firmware.koolshare.cn/Koolshare_RMerl_New_Gen_386/RT-AX86U/ 里选择7月30日发布的386固件进行下载
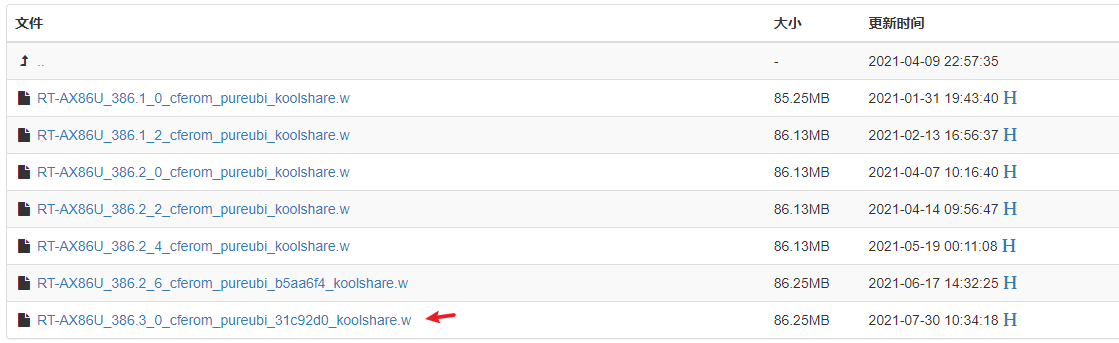
第二步:通过 router.asus.com 登录到路由器的后台,选择【系统管理】-> 【固件升级】, 上传下载的固件
静静等待固件升级完成。
第三步:登录路由器,选择最后的 【软件中心】,到【未安装】中安装 shellinabox,启动shell命令行,运行如下命令
1 | sed -i 's/\tdetect_package/\t# detect_package/g' /koolshare/scripts/ks_tar_install.sh |
第四步:离线安装插件。访问 https://github.com/hq450/fancyss ,点击 fancyss_hnd ,下载其中的 .tar.gz文件。接着,打开路由器后台,选择最后的软件中心的离线安装,上传该 tar.gz 文件,等待安装完成。
第五步: 配置第四步里安装的插件,即可实现更加自由的网页访问。
最后,感谢KoolShare论坛和GitHub的hq450。
hifiasm对HiFi PacBio进行组装
hifiasm是一个能有效利用PacBio HiFi测序技术,在分型组装图(pahsed assembly gprah)中可靠的表示单倍体信息的算法。
流程介绍
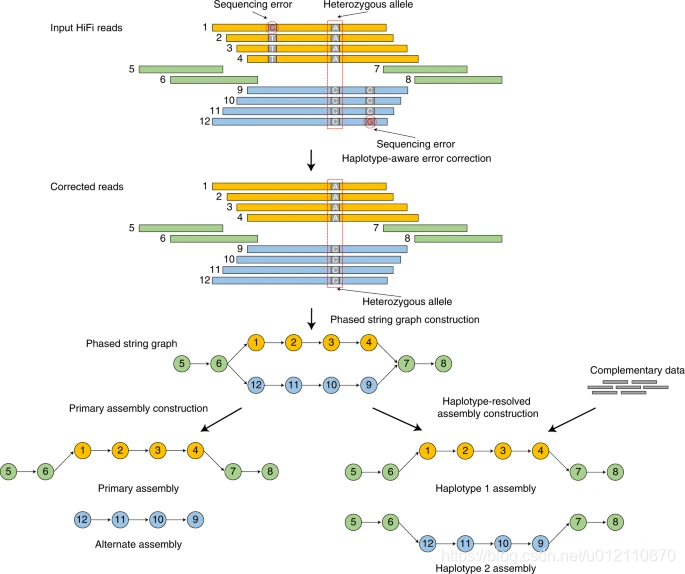
hifiasm的分析流程如下,主要分为3个阶段
第一阶段:通过所有序列的相互比对,对前在测序错误进行纠正。如果一个位置只存在两种碱基类型,且每个碱基类型至少有3条read支持,那么这个位置会被当作杂合位点,否则,视作测序错误,将被纠正。
第二阶段:根据序列之间的重叠关系,构建分型的字符串图(phased string graph)。其中调整朝向的序列作为顶点(vertex),一致重叠作为边(edge)。字符串图中的气泡(bubble)则是杂合位点。
第三阶段:如果没有额外的信息,hifiasm会随机选择气泡的一边构建primary assembly,另一边则是alternate assembly. 该策略和HiCanu,Falcon-Unzip一样。对于杂合基因组而言,由于存在一个以上的纯合haplotype,因此primary assembly可能还会包含haplotigs。HiCanu依赖于第三方的purge_dups, 而hifiasm内部实现了purge_dups算法的变种,简化了流程。如果有额外的信息,那么hifiasm就可以正确的对haplotype进行分型。
安装
hifiasm仅仅依赖 g++和zlib,以及git
1 | # 依赖g++和zlib |
通过源码编译的方式安装,需要将hifiasm移动到你的软件目录下,或者将hifiasm的路径加入到环境变量PATH中。
如果是 trio-binning模式,需要额外安装yak
1 | #source code |
案例展示
hifiasm的使用非常简洁明了,根据已有的数据分为,仅HiFi数据模式,有双亲二代测序的Trio-binning模式和有Hi-C数据的Hi-C模式。
仅有HiFi数据
最基本的用法,会得到两个部分分型的组装
1 | wget https://github.com/chhylp123/hifiasm/releases/download/v0.7/chr11-2M.fa.gz |
其中 -o定义输出文件的文件名前缀, -t是线程数
运行结束后生成的一系列文件中,我们只需要关注如下几项 (prefix表示前缀)
prefix.bp.r_utg.gfa: haplotype-resolved raw unitig graph,记录所有的单倍型信息prefix.bp.p_utg.gfa: 在raw unitig graph基础上过滤小的bubble,prefix.bp.p_ctg.gfa: 主要contig的assembly graphprefix.bp.hap1.p_ctg.gfa: haplotype1的部分分型的contig graphprefix.bp.hap2.p_ctg.gfa: haplotype2的部分分型的contig graph
如果并不需要部分分型的组装,而只想要primary和alternate的组装结果,可以在之前的命令的基础上,加上 --primary参数。
1 | hifiasm --primary -o test -t 32 chr11-2M.fa.gz 2> test.log2 |
由于hifiasm运行时会将步骤中纠错和相互比对的结果保存成 bin 文件,因此重新这一次运行速度会很快
primay模式下输出的文件和之前的类似,唯一的不同在于没有 bp
``prefix
.r_utg.gfa: haplotype-resolved raw unitig graph``prefix
.p_utg.gfa: haplotype-resolved processed unitig graph without small bubbles.``prefix
.p_ctg.gfa: assembly graph of primary contigs.``prefix
.a_ctg.gfa: assembly graph of alternate contigs.
我们关心的,可能就是 主要的contig,通过awk进行提取
1 | awk '/^S/{print ">"$2;print $3}' test.p_ctg.gfa > test.p_ctg.fa |
Trio-binning模式
如果测了双亲,则可以使用trio-binning方法进行更加可靠的分型。分为两步,先用yak统计k-mers, 然后用hifiasm进行组装
案例代码如下
1 | yak count -k31 -b37 -t16 -o pat.yak paternal.fq.gz |
输出的文章和之前类似,主要关心其中文件名带 dip 的输出gfa文件
prefix.dip.hap1.p_ctg.gfa: 完成分型的父源单倍体 contig图.prefix.dip.hap2.p_ctg.gfa: 完全分型的母源单倍体contig图.
整合Hi-C数据
由于Hi-C数据能够提供远距信息,因此也能用于单倍体分型。只需要加上两个参数, h1接受Hi-C的read1, h2 接受Hi-C的read2
1 | hifiasm -o NA12878.asm -t32 --h1 read1.fq.gz --h2 read2.fq.gz HiFi-reads.fq.gz |
在该模式下,每个contig要么是来自于父亲,要么是来自于母亲。hifiasm会将同一来源的contig放在同一个组装中。需要注意的是,hifiasm未必能够处理好着丝粒附近的区域,另外hifiasm中Hi-C也不会用于进行scaffold。
输出结果中,我们重点关注其中名字带hic的文件
prefix.hic.p_ctg.gfa: 主要contig的组装图prefix.hic.hap1.p_ctg.gfa: 完全分型的haplotype1的contig图prefix.hic.hap2.p_ctg.gfa: 完全分型的haplotype2的contig图prefix.hic.a_ctg.gfa: 如果设置了--primary参数,还会输出该次要contig的组装图
日志和参数调整
绝大部分的时候,我们只需要使用默认参数即可得到相对比较好的结果。但是当默认参数无法达到自己的目的,那我们就需要检查日志信息,阅读相关参数从而优化结果。
日志信息主要分为三项
k-mer图: 纯合样本只有一个peak,杂合样本则会有2个peak。
纯合峰的覆盖度:
[M::purge_dups] homozygous read coverage threshold: X, 一般会由hifiasm自动推断。杂合/纯合碱基数目(Hi-C模式): 在Hi-C模式下,如果纯合的碱基数超过杂合碱基数,那么hifiasm就不容易找对纯合read的所在峰。
对于日志信息,我们最主要关注的就是k-mer图,从而判断hifiasm是否能够正确的找到纯合峰,杂合峰的所在位置。如果hifiasm没有找对纯合峰所在的位置,就需要我们根据k-mer图手动指定 --hom-cov。
对于一个组装结果,最直接的评估标准就是基因组大小是否符合预期,分型的两套基因组是否相差不大,序列是否足够长,是否存在错误组装的情况。
如果基因组大小不符合预期,一般都是hifiasm找错了纯合峰的位置,我们需要手动指定 --hom-cov;如果分型的两套基因组差别过大,则通过降低 -s 调整。如果序列不够长,片段化明显,则可以尝试增加 -D 和 -N, 虽然会增加运行时间,但是会提高重复区域的分辨率。如果后续的Hi-C,或者BioNano发现hifiasm组装结果有比较多错误组装,则可以适当降低 --purge-max, -s和 -O。或者设置 -u 关闭post-join 步骤,hifiasm通过该步骤提高组装的连续性。
参考资料
https://github.com/chhylp123/hifiasm
在windows下编译bwa和samtools
通过在Windows上配置类Unix环境(MSYS2)里的操作,我们成功的在Windows里拥有了一个类Unix的环境,后续,我们就可以在这里面编译一些软件,这里以bwa和samtools为例
以下是一些必须的依赖项
- zlib-devel
- git
- make
- gcc
用pacman安装依赖项,如zlib-devel和git
1 | pacman -S zlib-devel |
如果连接git速度有问题,可以尝试按照如下方法配置镜像
1 | git config --global url."https://hub.fastgit.org/".insteadOf "https://github.com/" |
编译bwa
bwa只依赖于zlib,因此会很顺利
1 | git clone https://github.com/lh3/bwa |
编译htslib
samtools依赖于htslib,因此需要先编译htslib
1 | paman -S libbz2-devel liblzma-devel |
编译samtools
已知我不需要用到samtools 的 tview功能,则可以设置 --without-curses
1 | git clone https://github.com/samtools/samtools |
如果需要直接在cmd中调用msys2编译的程序,需要将 MSYS2 安装目录下的 usr/bin添加到Windows的环境变量中Path,参考在Windows上配置类Unix环境(MSYS2)里的操作
可能报错:
1 | make: *** 没有规则可制作目标“../htslib/htscodecs/htscodecs/arith_dynamic.c”,由“../htslib/hts-object-files” 需求。 停止。 |
原因: htslib没有克隆完全。
在Windows上配置类Unix环境(MSYS2)
MSYS2通过精心整合一些已有的工具(如gcc, qt),使得在Windows上也能使用类unix环境,调用类unix开发的软件。
它的官方地址为: https://www.msys2.org/
Cygwin尝试提供一个POSIX兼容环境,使得原本能在类unix环境里运行的软件,不需要经过太多修改就能直接在Windows上使用。它的特点是大而全,而MSYS2基于Cygwin提供优秀环境,使用了优秀的包管理工具pacman,更倾向于尝试提供一个构建原生Windows软件的环境。
安装步骤
第一步:在官网上下载MSYS2的安装包
e地址: https://github.com/msys2/msys2-installer/releases/download/2021-06-04/msys2-x86_64-20210604.exe
第二步: 运行MSYS2的安装程序,注意记录的你安装路径,我是修改了默认的安装路径为 D:\MSYS2

安装成功之后,你的菜单栏里就有了一个MSYS2的启动方式
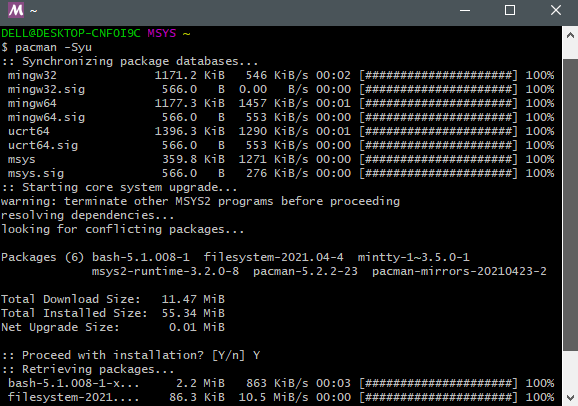
第三步: 更新pacman的包数据库
1 | pacman -Syu |
安装结束后,会关闭窗口,之后重新打开, 继续更新剩余的基础包
1 | pacman -Su |
第四步: 安装mingw-w64 GCC编译工具套装
1 | pacman -S --needed base-devel mingw-w64-x86_64-toolchain |
中间会有一些选项,全部enter默认即可。
其他
在CMD中调用MSYS2编译程序
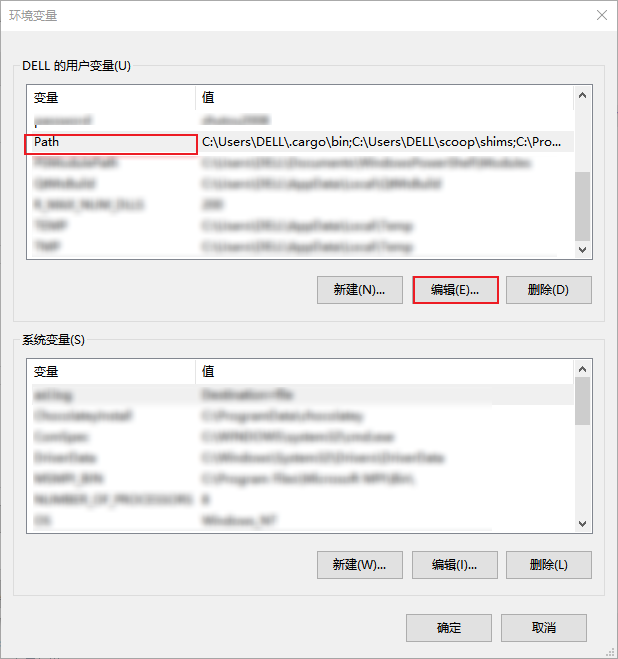
为了能够在cmd中使用MSYS2编译的软件,我们需要在环境变量中增加相应的路径,否则会出现如下报错
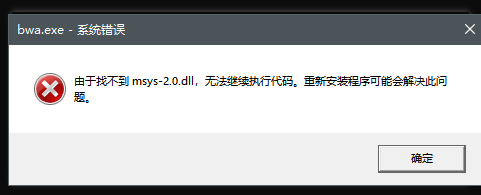
我安装的软件在D盘,因此我添加的路径是 D:\msys64\usr\bin
Pacman包管理
Pacman的常见操作
安装
1 | pacman -S <package_names|package_groups> |
移除
1 | pacman -R <package_names|package_groups> |
搜索
1 | pacman -Ss <name_pattern> |
- package_names: 包的名字
- package_groups: 包的所在组
- name_pattern: 包的可能命名