The operation couldn’t be completed. Unable to locate a Java Runtime. Please visit http://www.java.com for information on installing Java.
错误: package or namespace load failed for ‘rJava’: loadNamespace()里算'rJava'时.onLoad失败了,详细内容: 调用: fun(libname, pkgname) 错误: JVM could not be found 此外: Warning messages: 1: In system("/usr/libexec/java_home", intern =TRUE): 运行命令'/usr/libexec/java_home'的状态是1 2: In fun(libname, pkgname): Cannot find JVM library 'NA/lib/server/libjvm.dylib' Install Java and/or check JAVA_HOME (ifin doubt, do NOT set it, it will be detected)
> library(rgl) Error in dyn.load(dynlib <- getDynlib(dir)): 无法载入共享目标对象‘/Library/Frameworks/R.framework/Versions/4.1-arm64/Resources/library/rgl/libs/rgl.so’:: dlopen(/Library/Frameworks/R.framework/Versions/4.1-arm64/Resources/library/rgl/libs/rgl.so,0x0006): Library not loaded:/opt/X11/lib/libGLU.1.dylib Referenced from:/Library/Frameworks/R.framework/Versions/4.1-arm64/Resources/library/rgl/libs/rgl.so Reason: tried:'/opt/X11/lib/libGLU.1.dylib'(no such file),'/usr/lib/libGLU.1.dylib'(no such file) 错误: package or namespace load failed for ‘export’: loadNamespace()里算'rgl'时.onLoad失败了,详细内容: 调用: rgl.init(initValue, onlyNULL) 错误: OpenGL is not available in this build 此外: Warning messages: 1: Loading rgl's DLL failed. This build of rgl depends on XQuartz, which failed to load. See the discussion in https://stackoverflow.com/a/66127391/2554330 2: Trying without OpenGL...
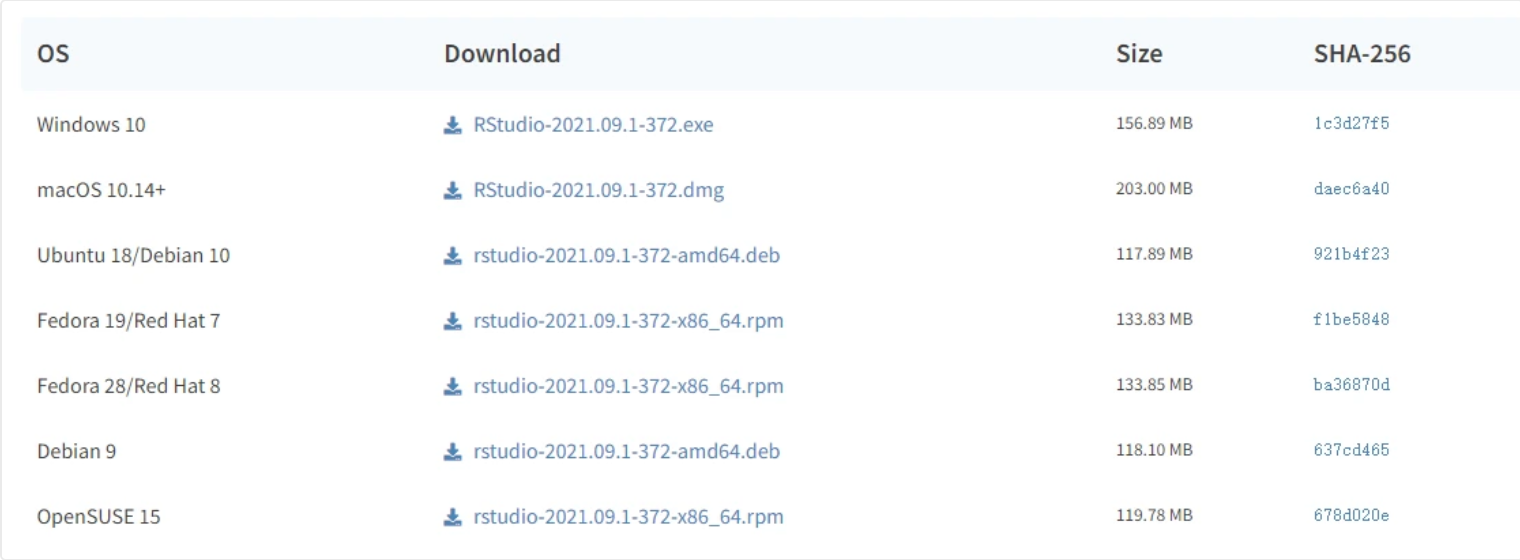
wget http://research-pub.gene.com/gmap/src/gmap-gsnap-2018-07-04.tar.gz tar xf gmap-gsnap-2018-07-04.tar.gz cd gmap-2018-07-04/ ./configure --prefix=$HOME/opt/biosoft/gmap make -j 20
GMAP: a genomic mapping and alignment program for mRNA and EST sequences Bioinformatics 2005 21:1859-1875 AbstractFull Text, Thomas D. Wu and Colin K. Watanabe
Fast and SNP-tolerant detection of complex variants and splicing in short reads Bioinformatics 2010 26:873-881 AbstractFull Text, Thomas D. Wu and Serban Nacu