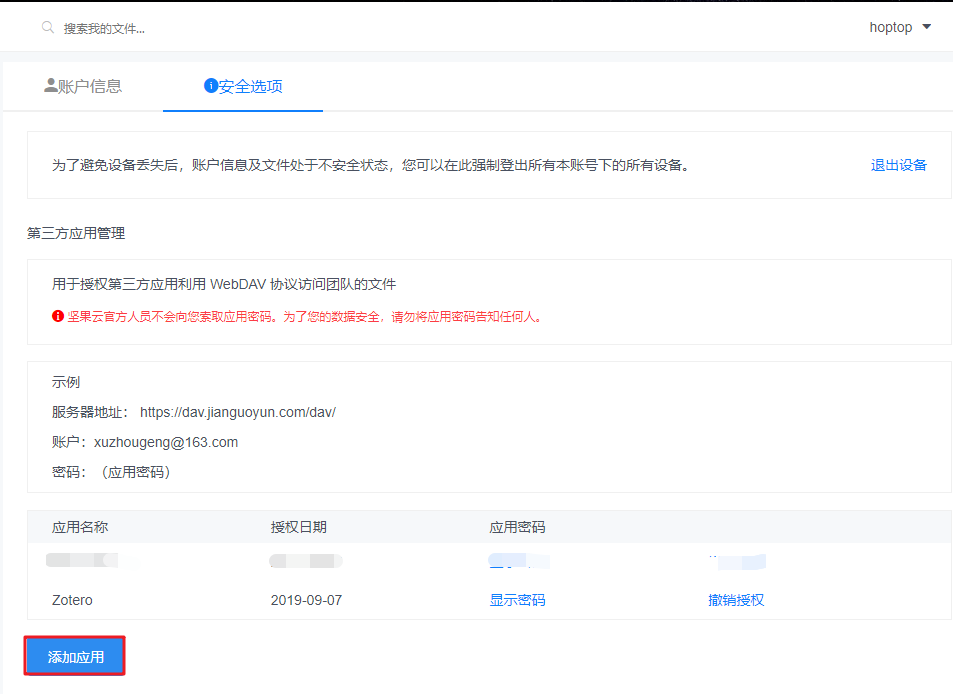
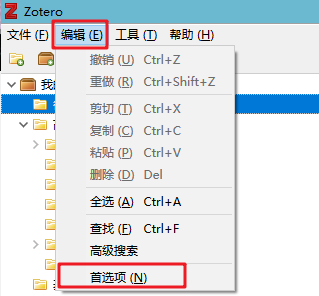
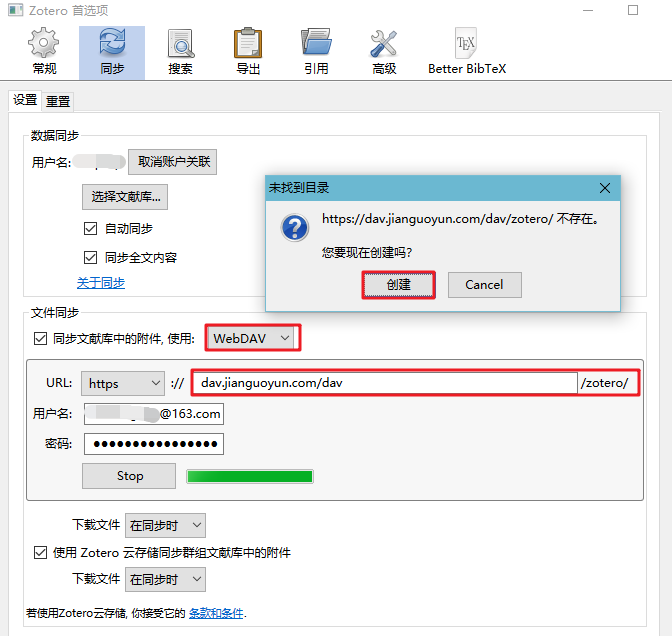
同一个R包,不同的安装界面
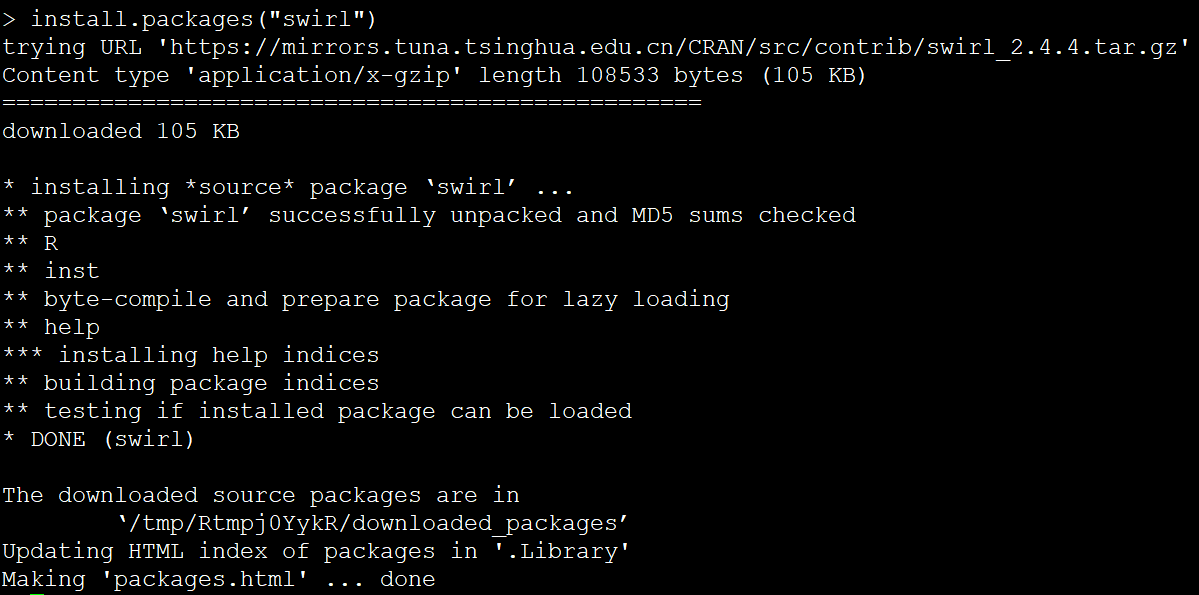
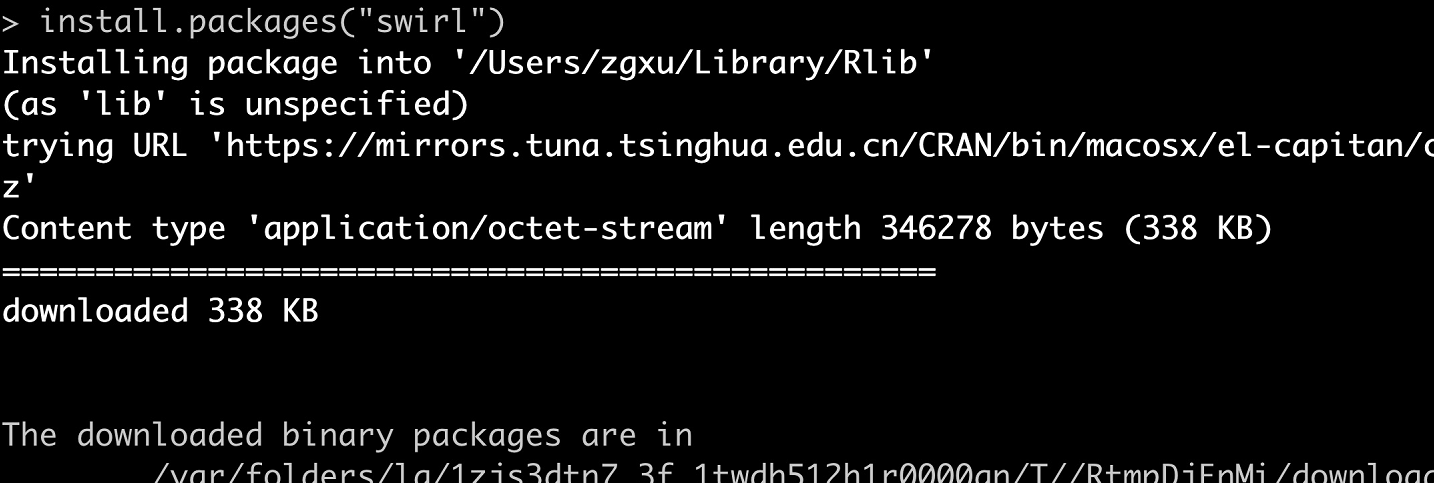
长久以来,我用iPad读文献的操作一直是下面这几步
每次插连接线都感觉自己太过原始。后来有了有了一部iPhoen手机,于是就变成了
手动清理是在是蠢。好在后来有了坚果云,就变成了
但是GoodReader这个软件只能作为一个阅读工具,它的PDF编辑功能太弱, 也不适合作为一个笔记工具
现在知道了PaperShip和MarginNote 3,于是就是
不过PaperShip这个软件存在一些问题,会出现文件无法同步的问题,只能期待有更好的App了。感谢坚果云,目前PaperShip能够顺利进行同步了。
只要打开坚果云官网,登录坚果云账号,在zotero文件夹新建空白的lastsync.txt文件,务必注意的是,必须使用坚果云自带的新建文件工具来新建lastsync.txt文件,不能通过手动上传的方式。
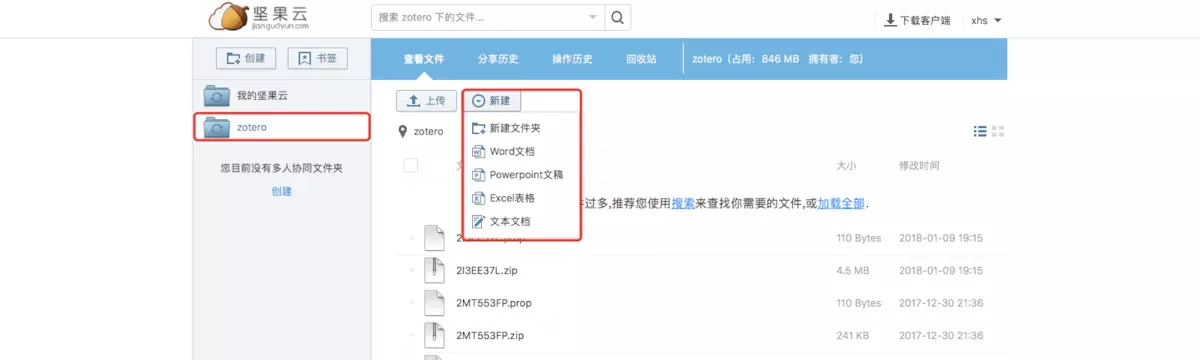
接着,重新在Zotero客户端或者Papership验证服务器即可
如果我想看的是一本书,那么就是把书放到坚果云中,用MarginNote 3打开坚果云里的书做笔记。
还有一个解决方案,是ZotFile这款插件。我们只需要新建一个坚果云同步文件夹,Tablet,并在Zotfile的插件中加入该选项。
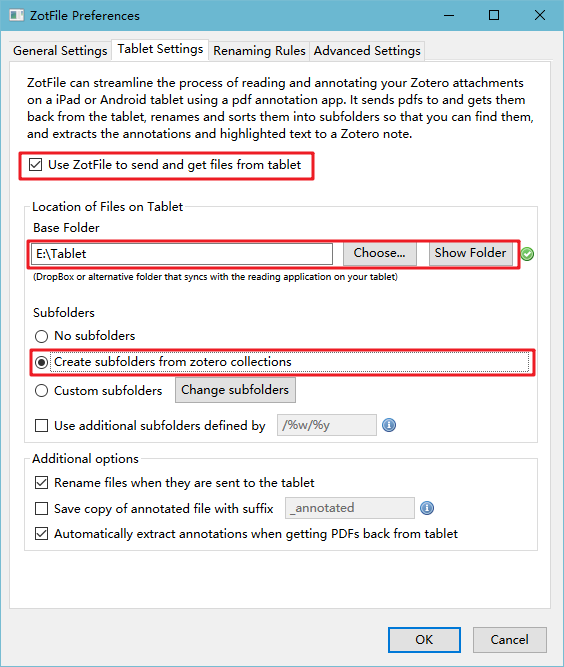
此时在Zotero处就会多出两个层次,存放送去批注的文件,以及批注完成的文件。
我们将要阅读文献右击,在Manage Attachment 中选择 Send to Tablet
因为我在GoodReader上设置坚果云中的Tablet作为同步服务器,因此就可以在GoodReader对文献进行批注。批注完成后,在Tablet Files(modified)中选择批注完成的文件,右击,在Manage Attachment 中选择 Get From Tablet
感觉这个方法也很不错,毕竟你回一次寝室看的文献也不会有那么多。
以ext4文件系统为例,设计的时候分为4个部分
df根据该信息进行统计磁盘占用ls -i获取i节点信息由于ls -l获取的是i节点记录的数据使用的数据块个数,而du则是通过i节点获取实际大小, 所以ls -l和du显示的数据大小不同。
RAID全称是Redundant Array of Independent Disks,也就是磁盘阵列,通过整合多块硬盘从而提升服务器数据的安全性,以及提高数据处理时的I/O性能。
RAID目前常用的是RAID5, 至少需要3块硬盘,其中一块硬盘用于奇偶校验,保证数据安全,其余硬盘同时读写,提高性能。此外,你还需要知道最原始的是RAID0,同时将数据读写到所有硬盘里,速度就变成了原来的N倍。RAID1至少需要两块盘,其中一块硬盘是另外硬盘的镜像。它不提高读写效率,只提高了数据安全性。RAID10是RAID0和RAID1的组合。
目前的服务器都配备了硬件RAID卡,因此在为服务器增加或更换硬盘时,需要格外注意,
fdisk只能对不多于2TB的硬盘进行分区
1 | fdisk /dev/sdb |
假如你的硬盘大于2TB,那么会输出如下信息
1 | Welcome to fdisk (util-linux 2.23.2). |
提示信息中的警告中,就建议”Use parted(1) and GUID partition table format (GPT).”
因此,对于大于2TB的硬盘就需要用parted进行分区
1 | parted /dev/sdb |
输出信息如下
1 | GNU Parted 3.1 |
创建新的GPT标签,例如
1 | mklabel gpt |
设置单位
1 | unit TB |
创建分区, 比如我将原来的10T分成2TB和8TB
1 | # mkpart PART-TYPE [FS-TYPE] START END |
查看分区表
1 |
输出如下
1 |
|
退出
1 | quit |
此时会提示”Information: You may need to update /etc/fstab.” /etc/fstab用于设置开机硬盘自动挂载。如果硬盘被拔走了,而/etc/fstab没有修改,那么会就提示进行修复模式。
在挂载硬盘之前,需要先对磁盘进行格式化。使用的命令为mkfs, 使用-t指定文件系统,或者用mkfs.xxx,其中xxx就是对应的文件系统。文件系统有如下几类
目前最流行的是ext4和xfs,足够稳定。其中xfs是CentOS7之后的默认文件系统。
1 | mkfs.ext4 /dev/sdb1 |
之后用mount进行硬盘挂载,分别两种情况考虑
一种是新建一个文件路径,进行挂载。
1 | mkdir data2 |
另一种是挂载一个已有目录,比如说临时文件目录/tmp挂载到新的设备中。
第一步: 新建一个挂载点,将原有数据移动到该目录下
1 | mkdir /storage |
第二步: 删除原来的/tmp下内容
1 | rm -rf /tmp/* |
第三步: 重新挂载
1 | umount /dev/sdb1 |
和mount相关的文件如下
此外mount在挂载的时候还可以设置文件系统参数,例如是否支持磁盘配额,对应-o参数
第零步: 检查服务器是否具备RAID阵列卡,如果有,则需要先为硬盘做RAID。
第一步: 使用fdisk -l检查硬盘是否能被系统检测到
第二步(可选): 假如需要硬盘分区,则用fdisk/gdisk/parted对硬盘划分磁盘
第三步: 使用mkfs进行磁盘格式化,有如下几种可选,
第四步: 用mkdir新建一个目录,然后用mount将格式化的硬盘挂载到指定目录下。卸载硬盘,则是umout
第五步: 修改/etc/fstab 将硬盘在重启的时候自动挂载。注意: 如果硬盘不在了,则需要将对应行注释掉,否则会进入到emergency模式。
谈论到直系同源基因分析的时候,大部分教程都是介绍OrthoMCL,这是2003年发表的一个工具,目前的引用次数已经达到了3000多,但这个软件似乎在2013年之后就不在更新,而且安装时还需要用到MySQL(GitHub上有人尝试从MySQL转到sqlite)。
而OrthoFinder则是2015年出现的软件,目前已有400多引用。该软件持续更新,安装更加友好,因此我决定使用它来做直系同源基因的相关分析。
OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy提到,它的优点就是比其他的直系同源基因组的推断软件准确,并且速度还快。
此外他还能分析所提供物种的系统发育树,将基因树中的基因重复事件映射到物种树的分支上,还提供了一些比较基因组学中的统计结果。
OrthoFinder的分析过程分为如下几步:
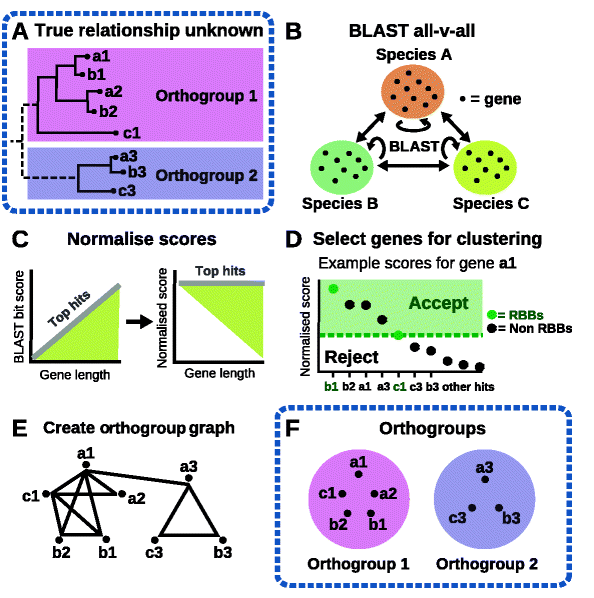
OrthoFinder2在OrthoFinder的基础上增加了物种系统发育树的构建,流程如下
基于Duplication-Loss-Coalescent 模型,有根基因树可以用来推断物种形成和基因复制事件,最后记录在统计信息中。
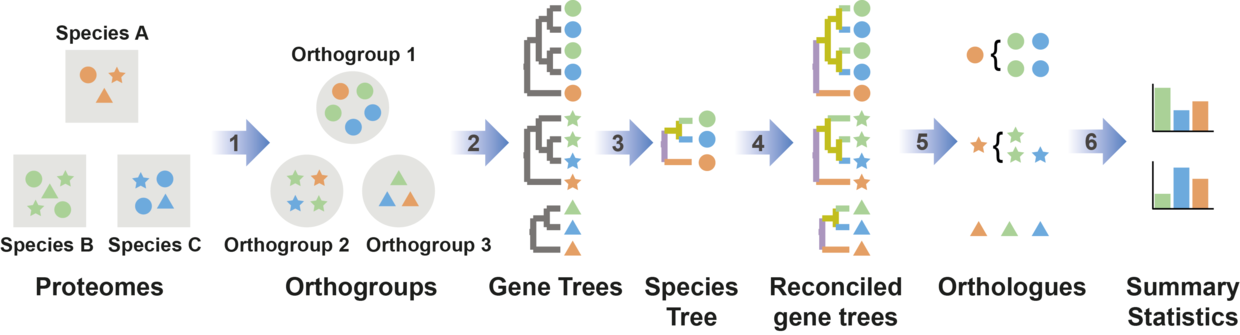
在解压缩的OrthoFinder文件目录下(安装见最后)有一个 ExampleData, 里面就是用于测试的数据集。
1 | orthofinder -f ExampleData -S mmseqs |
OrthoFinder的基本使用就是如此简单,而且最终效果也基本符合需求。
如果你想根据多序列联配(MSA)结果按照极大似然法构建系统发育树,那么你需要加上-M msa。这样结果会更加准确,但是代价就是运行时间会更久,这是因为OrthoFinder要做10,000 - 20,000个基因树的推断。
OrthoFinder默认用mafft进行多序列联配,用fasttree进行进化树推断。多序列联配软件还支持muscle, 进化树推断软件还支持iqtree, raxml-ng, raxml。例如参数可以设置为-M msa -A mafft -T raxml.
并行化参数: -t参数指定序列搜索时的线程数,-a指的是序列搜索后分析的CPU数。
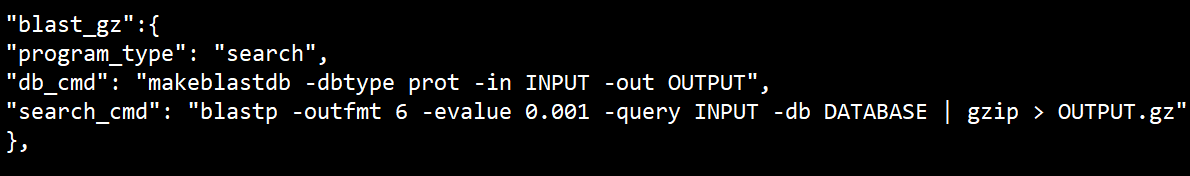
OrthoFinder提供了config.json可以调整不同软件的参数,如下是BLASTP。
OrthoFinder默认使用DendroBLAST发育树,也就是根据序列相似度推断进化关系。这是作者推荐的方法,在损失部分准确性的前提下提高了运算效率。当然你可以用-M msa从多序列比对的基础上进行基因树构建。如果你先用了默认的DendroBLAST,想测试下传统的MSA方法,那么也不需要重头运行,因为有一个-b参数可以在复用之前的比对结果。
在物种发育树的推断上,OrthoFinder使用STAG算法,利用所有进行构建系统发育树,而非单拷贝基因。此外当使用MSA方法进行系统发育树推断时,OrthoFinder为了保证有足够多的基因(大于100)用于分析,除了使用单拷贝基因外,还会挑选大部分是单拷贝基因的直系同源组。这些直系同源组的基因前后相连,用空缺字符表示缺失的基因,如果某一列存在多余50%的空缺字符,那么该列被剔除。最后基于用户指定的建树软件进行系统发育树构建。结果在”WorkingDirectory/SpeciesTree_unrooted.txt”
使用STRIDE算法从无根树中推断出有根树, 结果就是”SpeciesTree_rooted.txt”.
运行结束后,会在ExampleData里多出一个文件夹,Results_Feb14, 其中Feb14是我运行的日期
直系同源组相关结果文件,将不同的直系同源基因进行分组
直系同源相关文件,分析每个直系同源基因组里的直系同源基因之间关系,结果会在Orthologues_Feb14文件夹下,其中Feb14是日期
比较基因组学的相关结果文件:
STAG是一种从所有基因推测物种树的算法,不同于使用单拷贝的直系同源基因进行进化树构建。
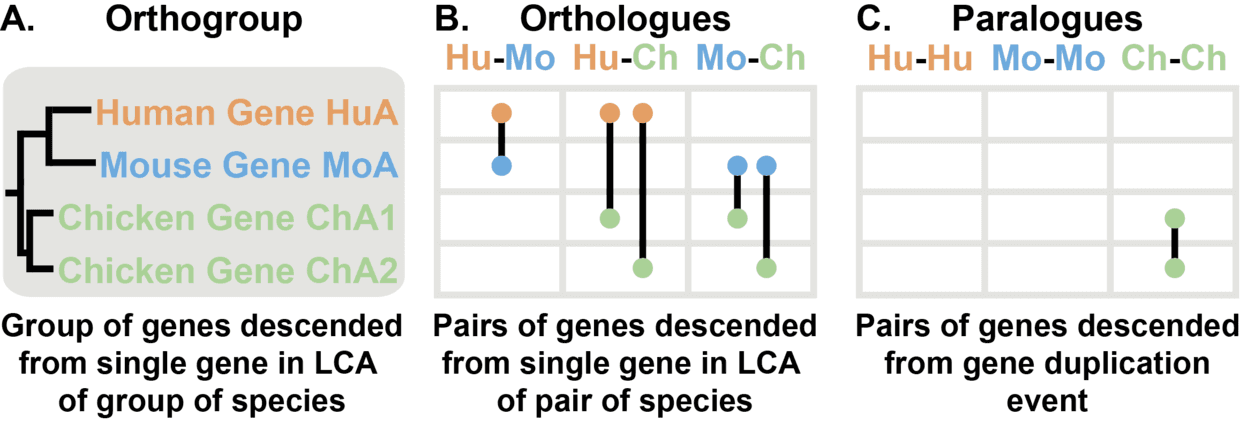
Orthogroups, Orthologs 和 Paralogs 这三个概念推荐看图理解。
OrthoFinder可以通过conda安装,建议为它新建一个虚拟环境
1 | conda create -n orthofinder orthofinder=2.2.7 |
你先得安装它的三个依赖工具: MCL, FastME, DIAMOND/MMseqs2/BLAST+
MCL有两种安装方式,最简单的就是用sudo apt-get install mcl, 但是对于大部分人可能没有root权限,因此这里用源代码编译。http://micans.org/mcl/
1 | wget https://www.micans.org/mcl/src/mcl-latest.tar.gz |
之后是MMseqs2, 一个蛋白搜索和聚类工具集,相关文章发表在NBT, NC上。GitHub地址为https://github.com/soedinglab/MMseqs2
1 | wget https://github.com/soedinglab/MMseqs2/releases/download/3-be8f6/MMseqs2-Linux-AVX2.tar.gz |
最后安装FastME, 这是一个基于距离的系统发育树推断软件。在http://www.atgc-montpellier.fr/fastme/binaries.php下载,上传到服务器
1 | tar xf fastme-2.1.5.tar.gz |
BLAST+可装可不装,推荐阅读这或许是我写的最全的BLAST教程
以上软件安装之后,都需要将其添加到环境变量中,才能被OrthoFinder调用。
之后在https://github.com/davidemms/OrthoFinder/releases 寻找最近的稳定版本下载到本地,例如OrthoFinder v2.2.7
1 | tar xzf OrthoFinder-2.2.7.tar.gz |
OrthoMCL是目前最常用的基因家族分析软件,从2013年发布2.0版本之后再也没有更新过,虽然它的安装过程复杂负责,但是依旧挡不住大家对他的喜好。
当然软件安装复杂是相对于之前,现在用Docker就可以轻松的安装和使用OrthoMCL。由于
1 | docker pull jasonkwan/orthomcl_docker |
由于该容器自带一个run_orthomcl.py, 只要你准备好输入数据和配置文件,就能够自动化进行分析.
其中run_orthomcl.py的参数有5个
其中-s和-e的1-4分别对应为
我们以一个具体的案例说明下。新建一个文件夹,例如说test, 里面是收集好的物种氨基酸序列。
1 | $ ls |
由于Docker版本的OrthoMCL使用DIAMOND进行blastp比对,因此一定要保证你的氨基酸序列中没有”.”,否则会报错。可以用seqkit grep过滤不合格的序列。
1 | seqkit grep -s -vrp '"\."' input.fa > output.faa |
之后创建一个fasta_table,放在test目录下。该文件分为两列,第一列是文件名,第二列是缩写。
1 | Alyrata.faa Aly |
准备配置文件orthomcl.config,同样放在test目录下
1 | dbVendor=mysql |
在test同级目录下运行如下命令
1 | docker run --privileged=true --rm -it --volume $PWD/test:/outdir:rw jasonkwan/orthomcl_docker:latest bash |
就会进入Docker的交互命令行,运行run_orthomcl.py
1 | cd outdir |
假如不希望进入交互命令行,那么需要按照下面的方法进行运行
1 | docker run --privileged=true --rm --volume $PWD/test:/outdir:rw jasonkwan/orthomcl_docker:latest /bin/bash -c "/tmp/.runconfig.sh && run_orthomcl.py --table_path /outdir/fasta_table --config_file /outdir/orthomcl.config --processors 32 " |
最终输出结果是groups.txt。下一个问题是,当你有了groups.txt后,下面能做什么分析呢?
按照对基因组学文章的整理,基本上就是下面两个
我正在找一篇文章尝试重现这个流程。
包括:blastn, blastp, blastx, tblastn, tblastx等. 使用conda安装即可。
1 | conda install -c bioconda blast |
BLAST数据库分为两类,核酸数据库和氨基酸数据库,可以用makeblastbd创建。可以用help参数简单看下说明。
1 | makeblastdb -help |
具体以拟南芥基因组作为案例,介绍使用方法:
注: 拟南芥的基因组可以在TAIR上下在,也可在ensemblgenomes下载。后者还可以下载其他植物的基因组
1 | 下载基因组 |
如果你从NCBI或者其他渠道下载了格式化过的数据库,那么可以用blastdbcmd去检索blast数据库,参数很多,常用就如下几个
使用案例
1 | 查看信息 |
还有其他有意思的参数,可以看帮助文件了解
安装nt/nr库需要先进行环境变量配置,在家目录下新建一个.ncbirc配置文件,然后添加如下内容
1 | ; 开始配置BLAST |
配置好之后,使用BLAST+自带的update_blastdb.pl脚本下载nr和nt等库文件(不建议下载序列文件,一是因为后者文件更大,二是因为可以从库文件中提取序列blastdbcmd -db nr -dbtype prot -entry all -outfmt “%f” -out nr.fa ,最主要是建库需要花费很长时间),直接运行下列命令即可自动下载。
1 | nohup time update_blastdb.pl nt nr > log & |
如果你不像通过update_blastdb.pl下载nr和nt等库文件,也可以是从ncbi上直接下载一系列nt/nr.xx.tar.gz文件,然后解压缩即可,后续还可以用update_blastdb.pl进行数据更新。
报错: 使用update_blastdb.pl更新和下载数据库时候出现模块未安装的问题。解决方法,首先用conda安装对应的模块,然后修改update_blastdb.pl的第一行,即shebang部分,以conda的perl替换,或者按照如下方法执行。
1 | perl `which update_blastdb.pl` |
下载过程中请确保网络状态良好,否则会出现Downloading nt.00.tar.gz...Unable to close datastream报错。
根据不同的需求,比如说你用的序列是氨基酸还是核苷酸,你要查找的数据是核甘酸还是氨基酸,选择合适的blast工具。不同需求的对应关系可以见下图(来自biostars handbook)
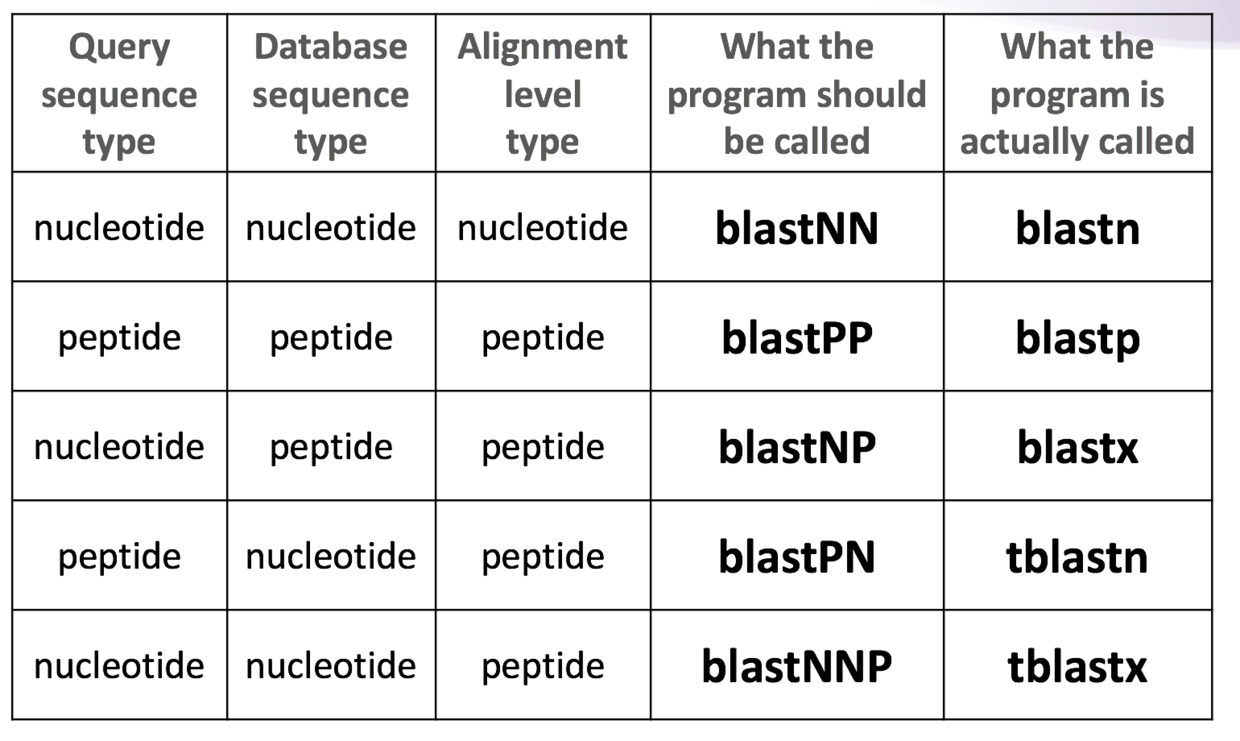
不同工具的应用范围虽然不同,但是基本参数都是一致的,学会blastn,基本上其他诸如blastp,blastx也都会了。
blastn的使用参数很多 blastn [-h] ,但是比较常用是如下几个
-query: 检索文件
-query_loc : 指定检索的位置
-strand: 搜索正义链还是反义链,还是都要
out : 输出文件
-remote: 可以用NCBI的远程数据库, 一般与 -db nr
-evalue 科学计数法,比如说1e3,定义期望值阈值。E值表明在随机的情况下,其它序列与目标序列相似度要大于这条显示的序列的可能性。 与S值有关,S值表示两序列的同源性,分值越高表明它们之间相似的程度越大
E值总结: 1.E值适合于有一定长度,而且复杂度不能太低的序列。2. 当E值小于10-5时,表明两序列有较高的同源性,而不是因为计算错误。3. 当E值小于10-6时,表时两序列的同源性非常高,几乎没有必要再做确认。
与联配计分相关参数: -gapopen,打开gap的代价;-gapextend, gap延伸的代价;-penalty:核酸错配的惩罚; -reward, 核酸正确匹配的奖励;
结果过滤:-perc_identity, 根据相似度
注 BLAST还提供一个task参数,感觉很有用的样子,好像会针对任务进行优化速度。
我随机找了一段序列进行检索
1 | echo '>test' > query.fa |
用的是blastn 检索核酸数据库。最简单的用法就是提供数据库所在位置和需要检索的序列文件
1 | blastn -db BLAST/TAIR10 -query query.fa |
以上是默认输出,blast的-outfmt选项提供个性化的选择。一共有18个选择,默认是0。
0 = Pairwise,
1 = Query-anchored showing identities,
2 = Query-anchored no identities,
3 = Flat query-anchored showing identities,
4 = Flat query-anchored no identities,
5 = BLAST XML,
6 = Tabular,
7 = Tabular with comment lines,
8 = Seqalign (Text ASN.1),
9 = Seqalign (Binary ASN.1),
10 = Comma-separated values,
11 = BLAST archive (ASN.1),
12 = Seqalign (JSON),
13 = Multiple-file BLAST JSON,
14 = Multiple-file BLAST XML2,
15 = Single-file BLAST JSON,
16 = Single-file BLAST XML2,
17 = Sequence Alignment/Map (SAM),
18 = Organism Report
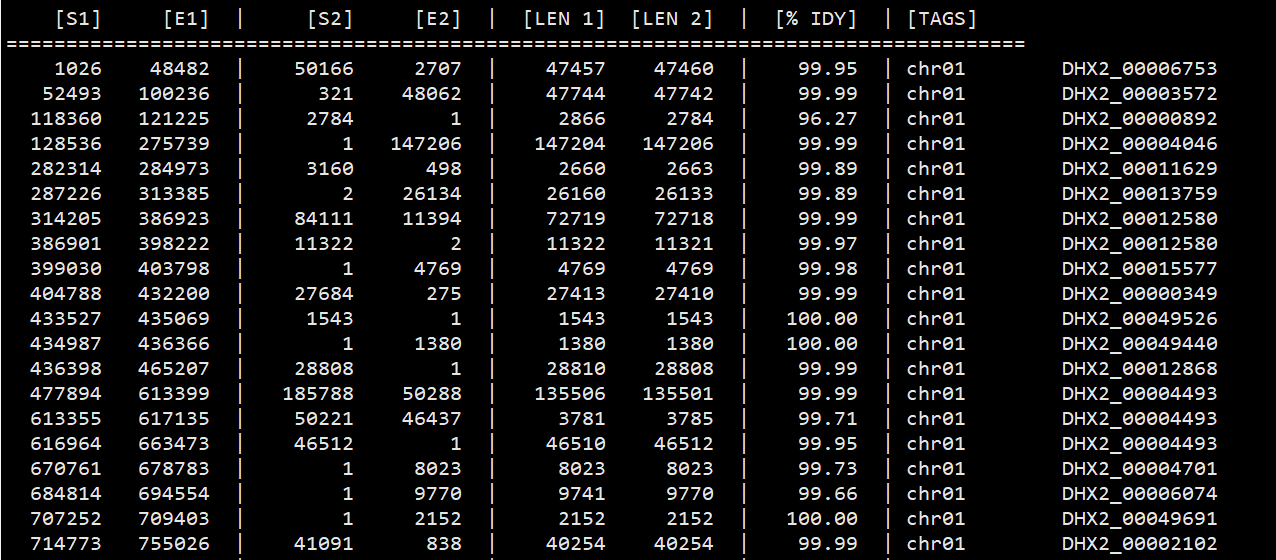
其中6,7,10,17可以自定输出格式。默认是
qaccver saccver pident length mismatch gapopen qstart qend sstart send evalue bitscore
| 简写 | 含义 |
|---|---|
| qaccver | 查询的AC版本(与此类似的还有qseqid,qgi,qacc,与序列命名有关) |
| saccver | 目标的AC版本(于此类似的还有sseqid,sallseqid,sgi,sacc,sallacc,也是序列命名相关) |
| pident | 完全匹配百分比 (响应的nident则是匹配数) |
| length | 联配长度(另外slen表示查询序列总长度,qlen表示目标序列总长度) |
| mismatch | 错配数目 |
| gapopen | gap的数目 |
| qstart | 查询序列起始 |
| qstart | 查询序列结束 |
| sstart | 目标序列起始 |
| send | 目标序列结束 |
| evalue | 期望值 |
| bitscore | Bit得分 |
| score | 原始得分 |
| AC: | accession |
以格式7为实例进行输出,并且对在线数据库进行检索
1 | blastn -task blastn -remote -db nr -query query.fa -outfmt 7 -out query.txt |
如果想输入序列,增加对应的格式qseq, sseq
1 | blastn -query query.fa -db nr -outfmt ' 6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send qseq sseq evalue bitscore' |
对已有序列进行注释时常见的best hit only模式命令行
1 | blastn -query gene.fa -out gene.blast.txt -task megablast -db nt -num_threads 12 -evalue 1e-10 -best_hit_score_edge 0.05 -best_hit_overhang 0.25 -outfmt "7 std stitle" -perc_identity 50 -max_target_seqs 1 |
仅仅看参数依旧无用,还需要知道BLAST的Best-Hits的过滤算法。假设一个序列存在两个match结果,A和B,无论A还是B,他们的HSP(High-scoring Segment Pair, 没有gap时的最高联配得分)一定要高于best_hit_overhang,否则被过滤。如果满足下列条件则保留A
曾经的BLAST安装后提供wwwblast用于构建本地的BLAST网页工具,但是BLAST+没有提供这个工具,好在BLAST足够出名,也就有人给它开发网页版工具。如viroBLAST和Sequenceserver, 目前来看似乎后者更受人欢迎。
有root安装起来非常的容易
1 | sudo apt install ruby ncbi-blast+ ruby-dev rubygems-integration npm |
数据库准备,前面步骤已经下载了拟南芥基因组的FASTA格式数据
1 | sequenceserver -d /到/之前/建立/BLAST/文件路径 |
然后就可以打开浏览器输入IP:端口号使用了。
开机启动:
新建一个/etc/systemd/system/sequenceserver.service文件,添加如下内容。注意修改ExecStart.
1 | [Unit] |
然后重新加载systemctl
1 | ## let systemd know about changed files |
nginx反向代理:我承认没有基本的nginx的知识根本搞不定这一步,所以我建议组内使用就不要折腾这个。简单的说就是在nginx的配置文件的server部分添加如下内容即可。
1 | location /sequenceserver/ { |
假如你有docker的话
1 | # ubuntu |
注意,你得在宿主机器上 /path-to-database-dir建好blast索引,或者用docker exec -it docker容器名 /bin/bash 进入到容器里,用sequenceserver -d db &运行
参考资料:http://www.sequenceserver.com/doc/
这是唐海宝老师GitHub上的JCVI工具的非官方说明书。
该工具集的功能非常多,但是教程资料目前看起来并不多,因此为了能让更多人用上那么好用的工具,我就一边探索,一边写教程
这一篇文章教大家如何利用JCVI里面的工具绘制点图,展现两个物种之间的共线性关系。
在分析之前,你需要从PhytozomeV11 下载A.thaliana和Alyrata的CDS序列,保证文件夹里有如下内容
1 | Alyrata_384_v2.1.cds.fa.gz Athaliana_167_TAIR10.cds.fa.gz |
我们在做CDS相互比对的时候只需要有每个基因最长的转录本即可,有两种方法可以实现
我用我写的一个脚本get_the_longest_transcripts.py提取每个基因的最长转录本,见 基因组共线性工具MCScanX使用说明
1 | zcat Alyrata_384_v2.1.gene.gff3.gz | python ~/scripts/python/get_the_longest_transcripts.py > aly_lst_gene.txt |
其中xxx_lst_gene.txt的格式如下, 第一列是基因名,第二列是mRNA编号,后面几列是位置信息。
1 | $ head ath_lst_gene.txt |
由于基因名和mRNA编号里有在提取CDS不需要的内容,因此要进行删除
1 | sed -i 's/\.v2\.1//g' aly_lst_gene.txt |
之后我们就可以根据第二列进行提取CDS
1 | seqkit grep -f <(cut -f 2 ath_lst_gene.txt ) Athaliana_167_TAIR10.cds.fa.gz > ath.cds |
提取的CDS编号里面也有一些不需要的内容,所以也要删除
1 | sed -i 's/\.t.*//' aly.cds |
此外还需要基因的位置信息的bed文件
1 | awk '{print $3"\t"$4"\t"$5"\t"$1"\t0\t"$6}' ath_lst_gene.txt | sort -k4,4V > ath.bed |
当然也可以参考「JCVI教程」如何基于编码序列或蛋白序列进行共线性分析来提取bed和cds序列,不需要用到我写的脚本。
1 | python -m jcvi.formats.gff bed --type=mRNA --key=Name Athaliana_167_TAIR10.gene.gff3.gz > ath.bed |
对bed文件中的基因进行去重
1 | python -m jcvi.formats.bed uniq ath.bed |
这一步会得到aly.uniq.bed和ath.uniq.bed, 我们将其覆盖原文件
1 | mv ath.uniq.bed ath.bed |
根据bed文件提取cds里的序列
1 | # Athaliana |
无论是哪种方法,请保证最后有以下四个文件
1 | $ ls ???.??? |
相对于上一步,这一步其实非常简单了
1 | makeblastdb -in ath.cds -out db/ath -dbtype nucl |
用jcvi.compara.blastfilter 对结果进行过滤
1 | python -m jcvi.compara.blastfilter --no_strip_names aly_ath.blast --sbed ath.bed --qbed aly.bed |
运行过程中有如下输出信息
1 | 19:59:15 [base] Load file `aly.bed` |
最后输出aly_ath.blast.filtered用于做图
1 | python -m jcvi.graphics.blastplot aly_ath.blast.filtered --sbed ath.bed --qbed aly.bed |
最后点图如下
在「JCVI教程」使用JCVI进行基因组共线性分析(上)还是有一个尴尬的事情,就是只用到两个物种,不能展示出JCVI画图的方便之处,因此这里参考https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version)的分析,只不过画图部分拓展下思路。
首先要运行如下代码获取目的数据
1 | python -m jcvi.apps.fetch phytozome Vvinifera,Ppersica,Tcacao |
然后按照教程的配置文件进行画图
layout文件内容如下
1 | # y, xstart, xend, rotation, color, label, va, bed |
seqids文件内容如下
1 | chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10,chr11,chr12,chr13,chr14,chr15,chr16,chr17,chr18,chr19 |
运行python -m jcvi.graphics.karyotype seqids layout会得到如下结果
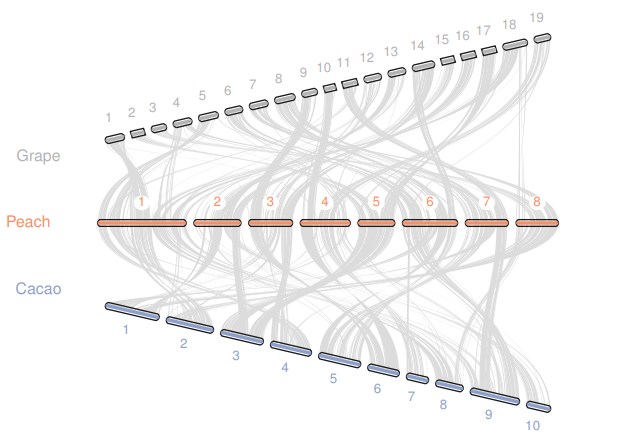
我就在思考一个问题如何让他形成一个三角形。经过一波三角运算和不断尝试,我定义了如下的layout
1 | # y, xstart, xend, rotation, color, label, va, bed |
那么效果怎么样呢?运行python -m jcvi.graphics.karyotype seqids layout吧
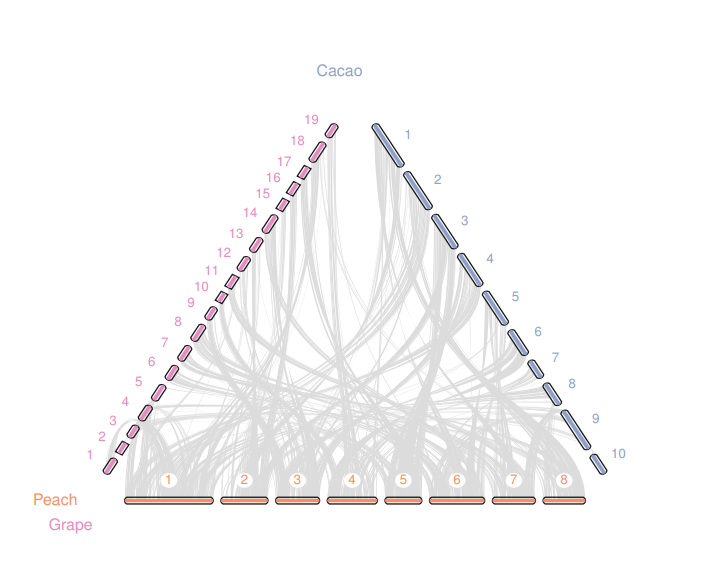
当然这里只展示了,grape和peach, peach和cacao之间的共线性,我又想着能不能加上grape和caocao呢?我尝试着运行下面的代码,
1 | python -m jcvi.compara.catalog ortholog grape cacao --cscore=.99 |
并继续修改了layout
1 | # y, xstart, xend, rotation, color, label, va, bed |
运行python -m jcvi.graphics.karyotype seqids layout会得到如下结果

我觉得给我一点时间,我也能用JCVI画出下面的图了
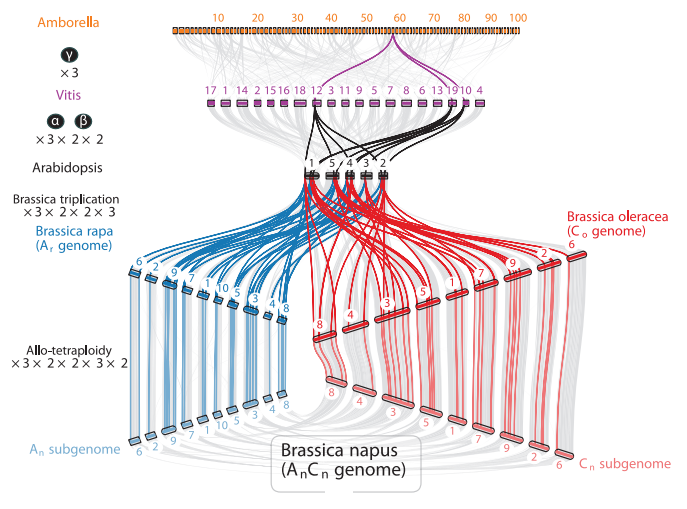
本教程借鉴https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version).
我们先从http://plants.ensembl.org/index.html选择两个物种做分析, 这里选择的就是前两个物种,也就是拟南芥和水稻(得亏没有小麦和玉米)

我们下载它的GFF文件,cdna序列和蛋白序列
1 | #Athaliana |
保证要有6个文件以便下游分析
1 | $ ls |
我们分析只需要用到每个基因最长的转录本就行,之前我用的是自己写的脚本,但其实我发现jcvi其实可以做到这件事情
先将gff转成bed格式,
1 | python -m jcvi.formats.gff bed --type=mRNA --key=transcript_id Arabidopsis_thaliana.TAIR10.44.gff3.gz > ath.bed |
然后将bed进行去重复
1 | python -m jcvi.formats.bed uniq ath.bed |
最后我们得到了ath.uniq.bed和osa.uniq.bed, 根据bed文件第4列就可以用于提取cds序列和蛋白序列。
1 | # Athaliana |
这里用到的seqkit建议学习,非常好用
下面使用python -m jcvi.compara.catalog ortholog进行共线性分析,这是一个非常行云流水的过程(除非你报错)
新建一个文件夹,方便在报错的时候,把全部都给删了,
1 | mkdir -p cds && cd cds |
运行代码
1 | python -m jcvi.compara.catalog ortholog --no_strip_names ath osa |
输出结果如下
1 | $ ls ath.osa.* |
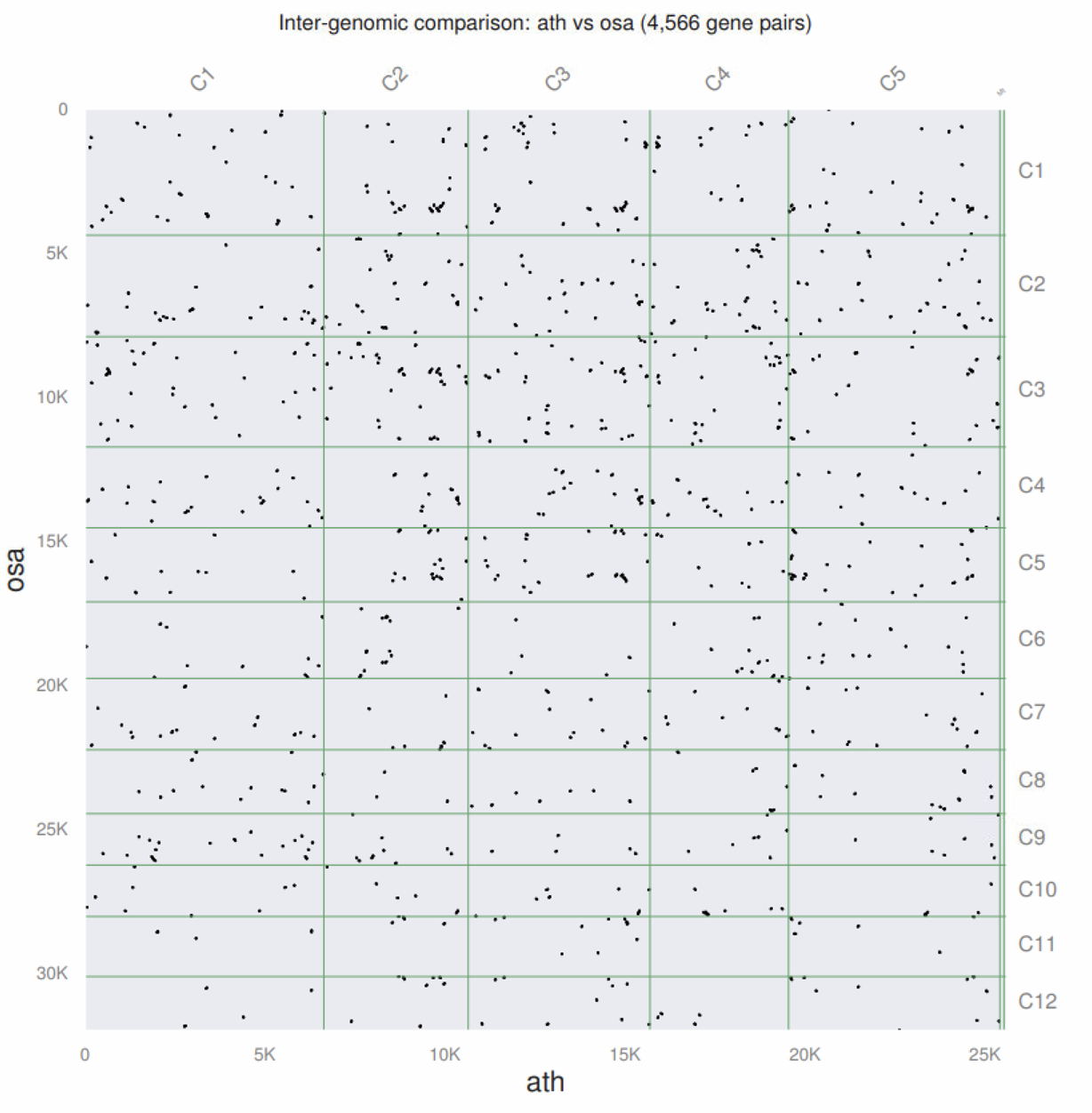
其中我们最感兴趣都是pdf结果,不出意外没啥共线性。

我们还可以用蛋白序列做共线性分析
1 | # 在之前输出cds,pep都文件夹操作 |
运行代码
1 | python -m jcvi.compara.catalog ortholog --dbtype prot --no_strip_names ath osa |
我之前以为他不可以基于蛋白序列分析,幸亏有人提醒。
你会发现这是一个自动化分析流程,我们只是提供了4个文件,它就完成了一些列事情。它生成的文件里除了PDF外,其他还有啥用呢?
anchors文件特别有用,之后会写一篇介绍如何利用他进行可视化,这里介绍它的格式。
1 | ### |
每个共线性区块以###进行分隔, 第一列是检索的基因,第二列是被检索的基因,第三列则是两个序列的BLAST的bit score,值越大可靠性越高。
用水稻和拟南芥进行了比较之后,发现后面基本上也没啥可以分析了。因此下面基于「JCVI教程」如何基于物种的CDS的blast结果绘制点图(dotplot)得到的cds和bed文件进行分析。
之前已经得到了如下四个文件
1 | ls ???.??? |
所以我们只要运行
1 | python -m jcvi.compara.catalog ortholog --no_strip_names aly ath |
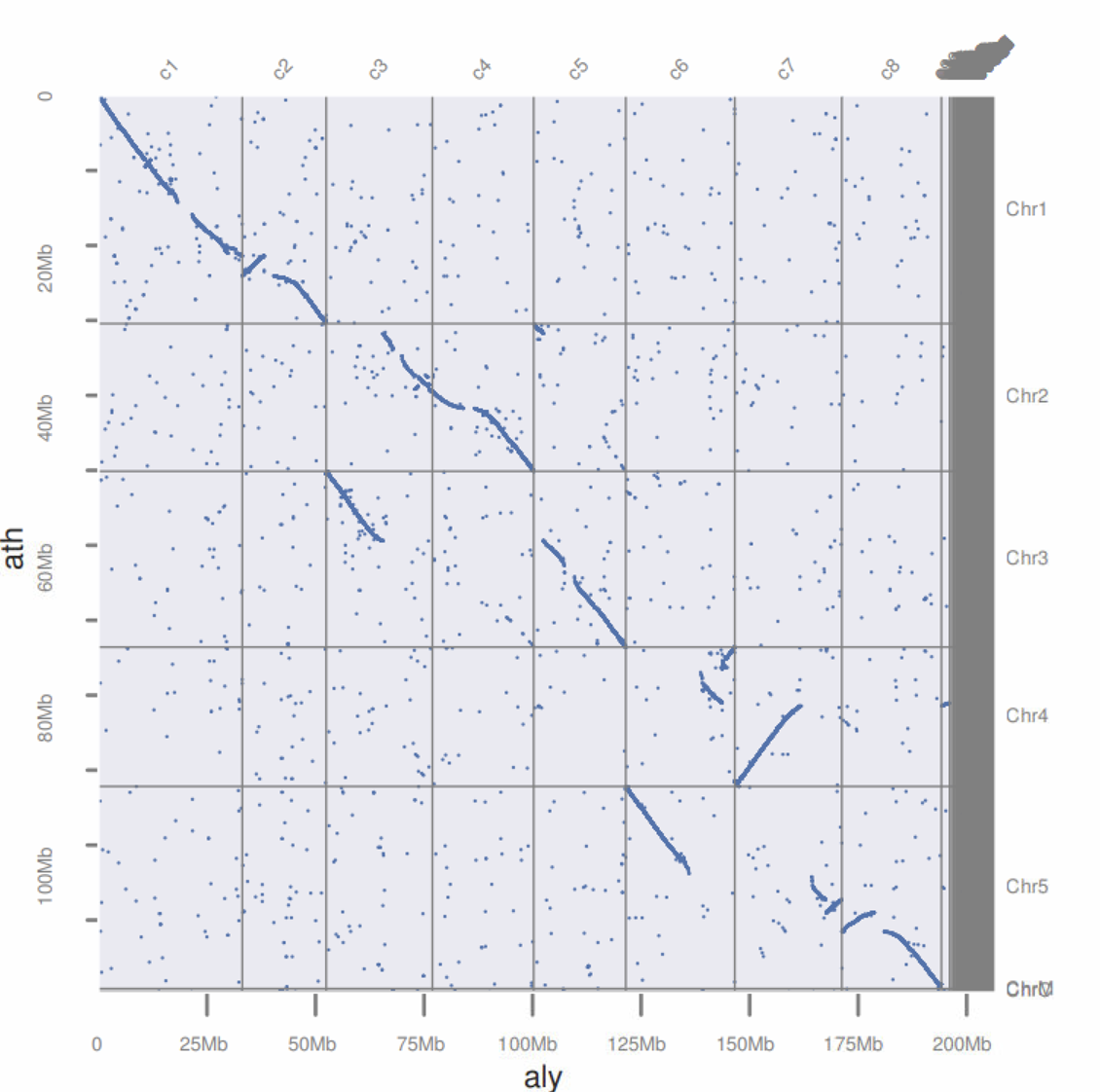
就得到了一个非常好看的点图
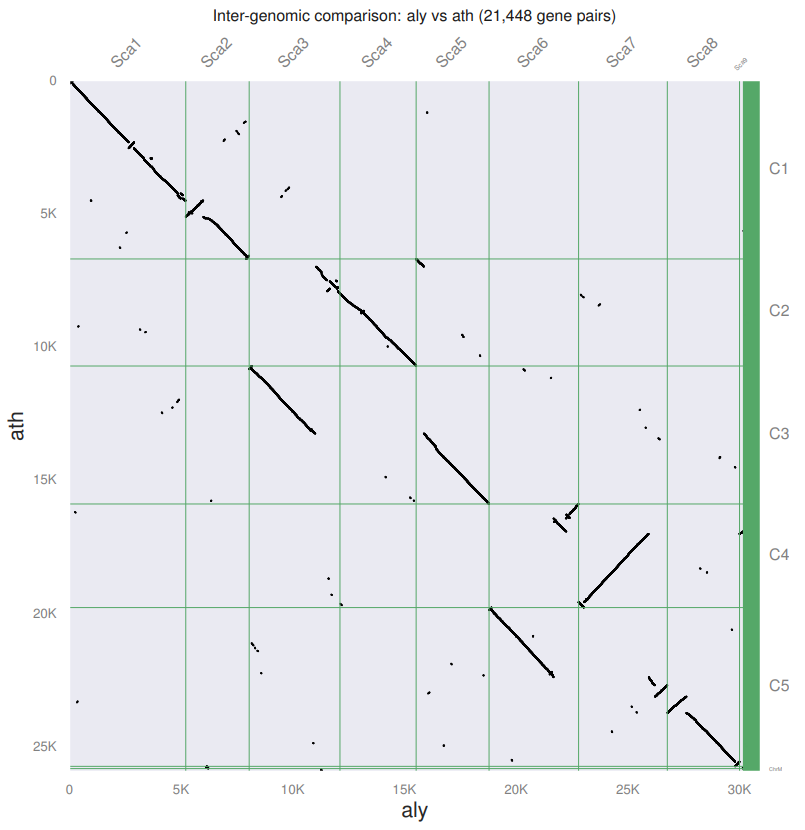
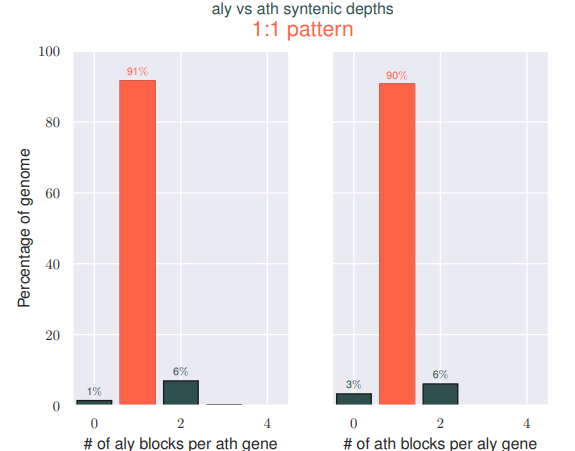
我们可以发现,都作为Arabidopsis属的两个物种,他们之间存在很高的同源性,并且同源区比例是1:1,
这其实和2011年的Nature Genetics上Alyrata的文章的结果是相似的,只不过他不是用点图进行展示
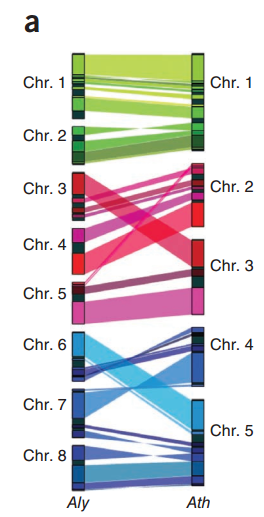
我们也可以用JCVI的画图模块实现这种效果,只不过还需要一点额外操作,创建如下三个文件
.anchors文件创建的更简化格式第一步,创建.simple文件
1 | python -m jcvi.compara.synteny screen --minspan=30 --simple aly.ath.anchors aly.ath.anchors.new |
第二步, 创建seqid文件,非常简单,就是需要展示的scaffold或染色体的编号
1 | scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8 |
第二步,创建layout文件,用于设置绘制的一些选项。
1 | # y, xstart, xend, rotation, color, label, va, bed |
注意, #edges下的每一行开头都不能有空格
最后运行下面的命令,会得到一个karyotype.pdf
1 | python -m jcvi.graphics.karyotype seqids layout |

如何让这个图垂直呢?(导入AI里就好了)