这部分内容是Extending ggplot2 的学习笔记,大部分内容都是原文的简单翻译。
所有的ggplot2对象都建立自”ggproto”这套面向对象编程系统,因此想要创建出自己的一套图层,而不是简单的对已有图层进行累加,那么就需要学习”ggproto”。
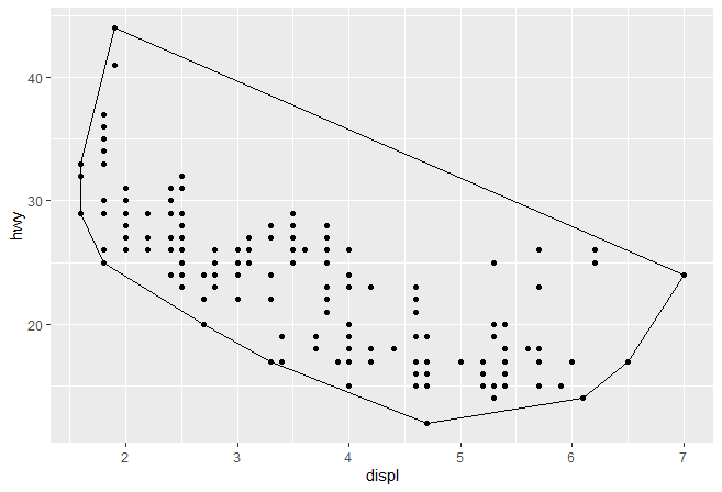
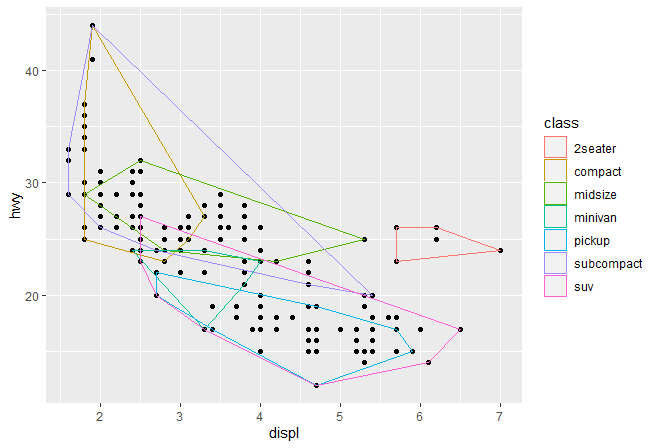
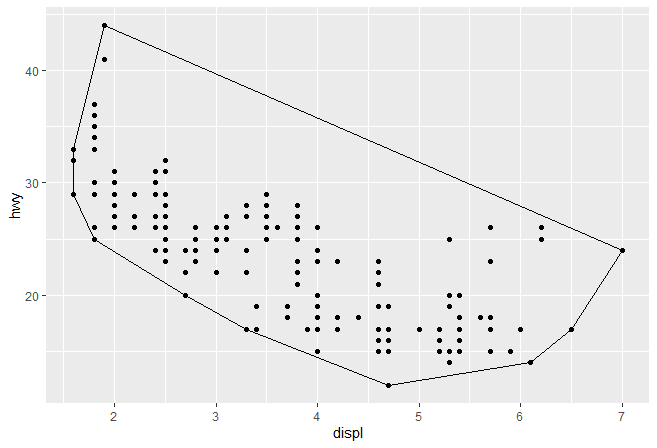
创建新的stat 最简单的stat 我们会从一个最简单的stat开始: 根据已有的一组点,用一个凸壳(convex hull)包围他。
第一步,我们创建一个继承自Stat的”ggproto”对象
1 2 3 4 5 6 7 StatChull <- ggproto( "StatChull" , Stat, compute_group = function ( data, scales) { data[ chull( data$ x, data$ y) , , drop= FALSE ] } , required_aes = c ( "x" , "y" ) )
在”ggproto”函数中,前两个是固定项,分别是类名 和继承的”ggproto”类。而后续内容则是和你继承的类相关,例如compute_group()方法负责计算,required_aes则列出哪些图形属性(aesthetics)必须要存在,这两个都继承自Stat,可以用?Stat查看更多信息。。
第二步,我们开始写一个图层 。由于历史设计原因,Hadley将其称作stat_()或geom_()。但实际上,Hadley认为layer_()可能更准确些,毕竟每一个图层都或多或少的有”stat”和”geom”。
所有的图层都遵循相同的格式 ,即你在function中声明默认参数,然后调用layer()函数,将...传递给params参数。在...的参数既可以是”geom”的参数(如果你要做一个stat封装),或者是”stat”的参数(如果你要做geom的封装),或者是将要设置的图形属性. layer()会小心的将不同的参数分开并确保它们存储在正确的位置:
1 2 3 4 5 6 7 8 9 10 stat_chull <- function ( mapping = NULL , data = NULL , geom = "polygon" , position = "identity" , na.rm = TRUE , show.legend = NA , inherit.aes = TRUE , ...) { layer( stat = StatChull, data = data, mapping = mapping, geom = geom, position = position, show.legend = show.legend, inherit.aes = inherit.aes, params = list ( na.rm = na.rm, ...) ) }
(注 , 在写R包的时候要注意用ggplot2::layer()或在命名空间中导入layer(), 否则会因找到函数而报错)
当我们写好了图层函数后,我们就可以尝试这个新的”stat”了
1 2 3 ggplot( mpg, aes( displ, hwy) ) + geom_point( ) + stat_chull( fill = NA , colour= "black" )
(后续我们会学习如何通过设置”geom”的默认值,来避免声明fill=NA)
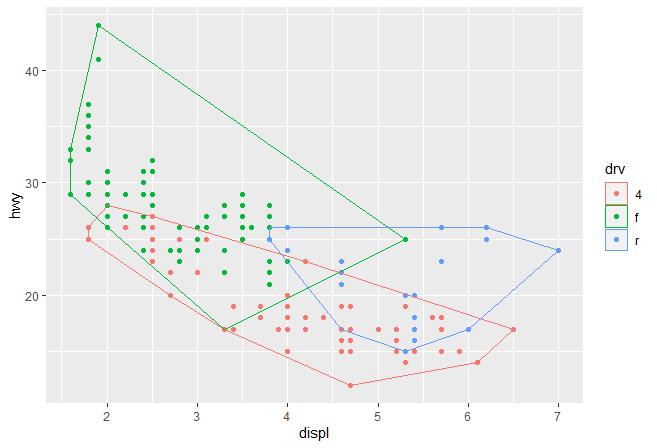
一旦我们构建了这种基本的对象,ggplot2将会给我们带来极大的自由。举个例子,ggplot2自动保留每组中不变的图形属性,也就是说你可以分组绘制一个凸壳:
1 2 3 ggplot( mpg, aes( displ, hwy, colour = drv) ) + geom_point( ) + stat_chull( fill = NA )
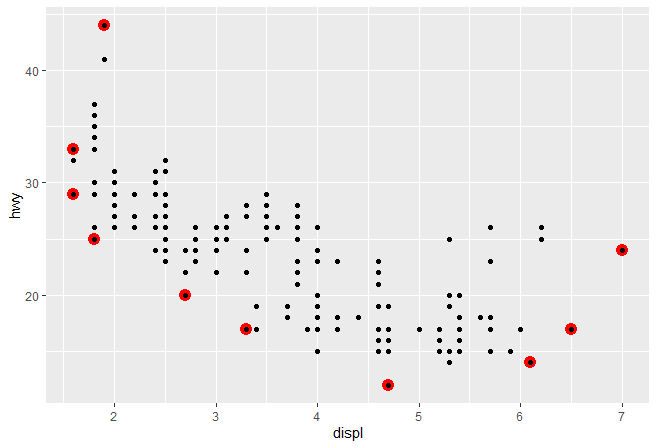
我们还可以覆盖默认的图层,来以不同的形式展现凸壳:
1 2 3 ggplot( mpg, aes( displ, hwy) ) + stat_chull( geom = "point" , size = 4 , colour = "red" ) + geom_point( )
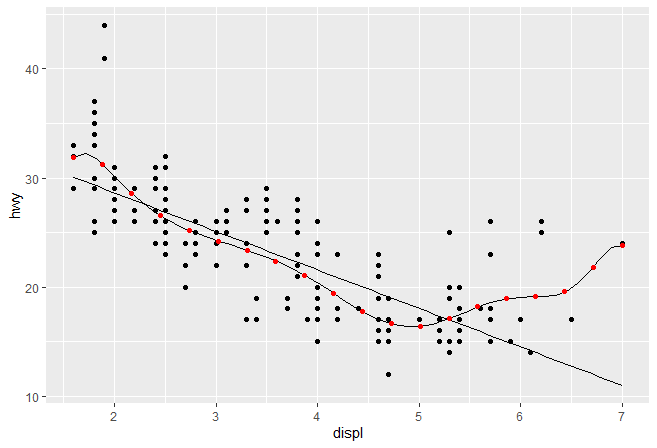
Stat参数 一个更加复杂的”stat”会做一些计算。我们可以通过实现一个简单版本的geom_smooth来了解。我们将会创建一个新的图层StatLm(继承自Stat)和一个的图层函数stat_lm():
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 StatLm <- ggproto( "StatLm" , Stat, required_aes = c ( "x" , "y" ) , compute_group = function ( data, scales) { rng <- range ( data$ x, nr.rm = TRUE ) grid <- data.frame( x = rng) mod <- lm( y ~ x, data = data) grid$ y <- predict( mod, newdata = grid) grid } ) stat_lm <- function ( mapping = NULL , data = NULL , geom = "line" , position = "identity" , na.rm = FALSE , show.legend = NA , inherit.aes = TRUE , ...) { layer( stat = StatLm, data = data, mapping = mapping, geom = geom, position = position, show.legend = show.legend, inherit.aes = inherit.aes, params = list ( na.rm = na.rm, ...) ) }
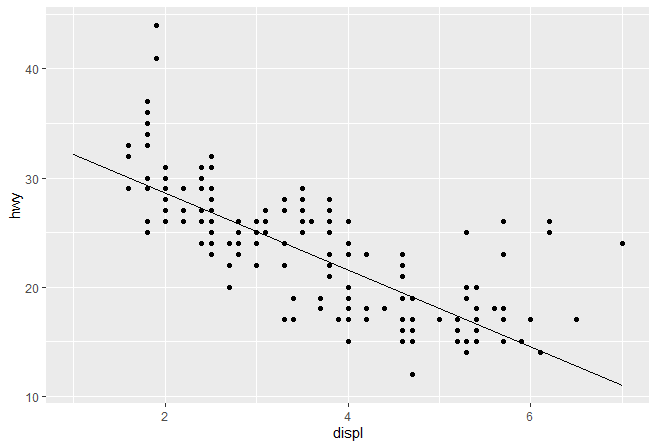
调用我们写的stat_lm()图形,检查下效果
1 2 3 ggplot( mpg, aes( displ, hwy) ) + geom_point( ) + stat_lm( )
StatLm缺少参数不太灵活,只能做单一线性拟合。最好是允许用户能够自由修改模型公式和创建图层所需要的数据量。为了实现这一需求,我们在compute_group()增加了一些参数,代码如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 StatLm2 <- ggproto( "StatLm2" , Stat, required_aes = c ( "x" , "y" ) , compute_group = function ( data, scales, params, n = 100 , formula = y ~ x) { rng <- range ( data$ x, na.rm = TRUE ) grid <- data.frame( x = seq( rng[ 1 ] , rng[ 2 ] , length = n) ) mod <- lm( formula, data = data) grid$ y <- predict( mod, newdata = grid) grid } ) stat_lm2 <- function ( mapping = NULL , data = NULL , geom = "line" , position = "identity" , na.rm = TRUE , show.legend = NA , inherit.aes = TRUE , n = 50 , formula = y ~ x, ...) { layer( stat = StatLm2, data = data, mapping = mapping, geom = geom, position = position, show.legend = show.legend, inherit.aes = inherit.aes, params = list ( n = n, formula = formula, na.rm = na.rm, ...) ) } ggplot( mpg, aes( displ, hwy) ) + geom_point( ) + stat_lm( ) + stat_lm2( formula = y ~ poly( x, 10 ) ) + stat_lm2( formula = y ~ poly( x, 10 ) , geom = "point" , colour = "red" , n = 20 )
我们并不需要显式在图层中包括新的参数,..会将这些参数放到合适的地方。但是你必须在文档中写出哪些参数是可以让用户调整的,以便用户知道他们的存在。举个一个简单的例子
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 stat_lm <- function ( mapping = NULL , data = NULL , geom = "line" , position = "identity" , na.rm = FALSE , show.legend = NA , inherit.aes = TRUE , n = 50 , formula = y ~ x, ...) { layer( stat = StatLm, data = data, mapping = mapping, geom = geom, position = position, show.legend = show.legend, inherit.aes = inherit.aes, params = list ( n = n, formula = formula, na.rm = na.rm, ...) ) }
上面代码中以#' 开头内容都是roxygon语法,其中@inheritParams ggplot2::stat_identity表示在最后输出的帮助文档中会继承stat_identity的参数说明。而@export则是将函数让用户可见,否则用户无法直接调用。
挑选参数 有些时候,你会发现部分运算是针对所有数据集进行,而非每个分组。比较好的方法就是挑选明智的默认值。例如,我们需要做密度预测,我们有理由为整个图形挑选一个带宽(bandwidth)。下面的”Stat”创建了stat_density()的变体,通过选择每组最优带宽的均值作为所有分组的带宽。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 StatDensityCommon <- ggproto( "StatDensityComon" , Stat, required_aes = "x" , setup_params = function ( data, params) { if ( ! is.null ( params$ bandwidth) ) return ( params) xs <- split( data$ x, data$ group) bws <- vapply( xs, bw.nrd0, numeric( 1 ) ) bw <- mean( bws) message( "Picking bandwidth of " , signif ( bw, 3 ) ) params$ bandwidth <- bw params } , compute_group = function ( data, scales, bandwidth = 1 ) { d <- density( data$ x, bw = bandwidth) data.frame( x = d$ x, y = d$ y) } ) stat_density_common <- function ( mapping = NULL , data = NULL , geom = "line" , position = "identity" , na.rm = FALSE , show.legend = NA , inherit.aes = TRUE , bandwidth = NULL , ...) { layer( stat = StatDensityCommon, data = data, mapping = mapping, geom = geom, position = position, show.legend = show.legend, inherit.aes = inherit.aes, params = list ( bandwidth = bandwidth, na.rm = na.rm, ...) ) } ggplot( mpg, aes( displ, colour = drv) ) + stat_density_common( )
作者推推荐用NULL作为默认值。如果你通过自动计算的方式挑选了重要参数,那么建议通过message()的形式告知用户(在答应浮点值参数时,用singif()可以只展示部分小数点)。
变量名和默认图形属性 这部分”stat”会阐述另外一个重要的点。当我们想要让当前”stat”对其他geoms更加有用时,我们应该返回一个变量,称之为”density”而不是”y”。之后,我们可以设置”default_aes”自动地将density映射到y, 这允许用户覆盖它从而使用不同的”geom”.
1 2 3 4 5 6 7 8 9 10 11 StatDensityCommon <- ggproto( "StatDentiy2" , Stat, required_aes = "x" , default_aes = aes( y = stat( density) ) , compute_group = function ( data, scales, bandwidth = 1 ) { d <- density( data$ x, bw= bandwidth) data.frame( x = d$ x , density= d$ y) } ) ggplot( mpg, aes( displ, drv, colour = stat( density) ) ) + stat_density_common( bandwidth = 1 , geom= "point" )
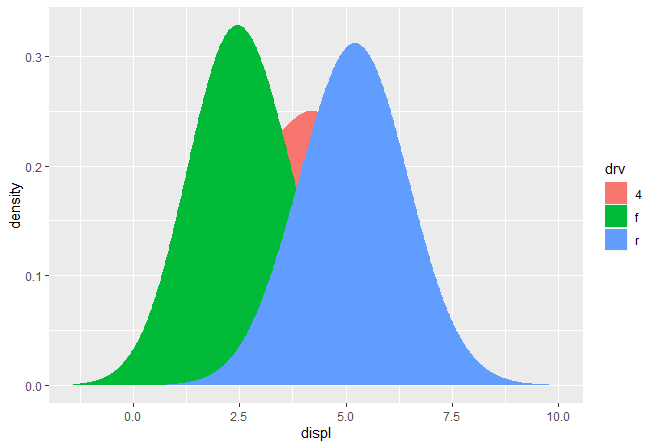
然而直接在stat中用area geom的结果可能和你想的不同。
1 2 ggplot( mpg, aes( displ, fill = drv) ) + stat_density_common( bandwidth = 1 , geom = "area" , position = "stack" )
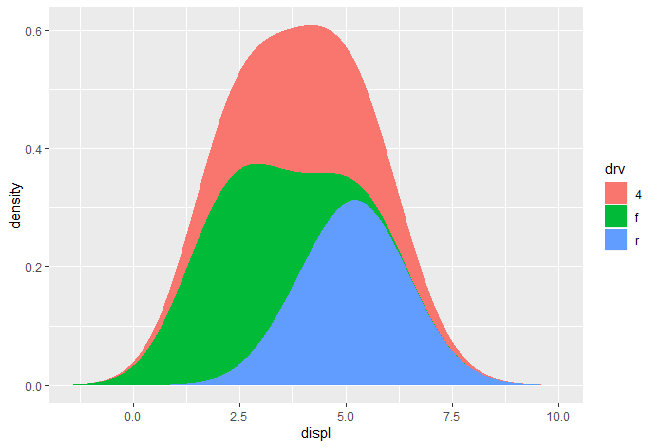
密度不是一个相互累加,而是单独计算,因此预测的x没有对齐。我们可以通过在setup_params()计算数据范围的方式解决该问题
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 StatDensityCommon <- ggproto( "StatDensityCommon" , Stat, required_aes = "x" , default_aes = aes( y = stat( density) ) , setup_params = function ( data, params) { min <- min ( data$ x) - 3 * params$ bandwidth max <- max ( data$ x) + 3 * params$ bandwidth list ( bandwidth = params$ bandwidth, min = min , max = max , na.rm = params$ na.rm ) } , compute_group = function ( data, scales, min , max , bandwidth = 1 ) { d <- density( data$ x, bw = bandwidth, from = min , to = max ) data.frame( x = d$ x, density = d$ y) } ) ggplot( mpg, aes( displ, fill = drv) ) + stat_density_common( bandwidth = 1 , geom = "area" , position = "stack" )
使用”raster”几何形状
1 2 ggplot( mpg, aes( displ, drv, fill = stat( density) ) ) + stat_density_common( bandwidth = 1 , geom = "raster" )
练习题
拓展stat_chull,使其能够计算alpha hull, 类似于alphahull. 新的”stat”能够接受alpha做为参数
修改最终版本的StatDensityComon, 使其能够接受用户定义的min和max. 你需要同时修改layer函数和compute_group()方法
将StatLm和ggplot2::StatSmooth对比。是什么差异使得StatSmooth比StatLm更加复杂。
创建新的geom 相对于创建新的”stat”, 创建新的”geom”会将难一些,因为这需要你懂得一些grid知识。因为ggplot2基于grid,所以你得要学一些用grid绘图的知识。如果你真的打算学习如何新增一个新的”geom”,Hadley推荐你买Paul Murrell所著的R绘图系统 。里面介绍所有和用”grid”绘图相关的知识。
一个简单的geom 让我们先从一个简单的案例入手,尝试实现一个类似于geom_point()的图层
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 GeomSimplePoint <- ggproto( "GeomSimplePoint" , Geom, required_aes = c ( "x" , "y" ) , default_aes = aes( shape = 19 , size = 0.1 , colour = "black" ) , draw_key = draw_key_point, draw_panel = function ( data, panel_params, coord) { coords <- coord$ transform( data, panel_params) grid:: pointsGrob( coords$ x, coords$ y, pch = coords$ shape, size = unit( coords$ size, "char" ) , gp = grid:: gpar( col= coords$ colour) ) } ) geom_simple_point <- function ( mapping = NULL , data = NULL , stat = "identity" , position = "identity" , na.rm = FALSE , show.legend = NA , inherit.aes = TRUE , ...) { layer( geom = GeomSimplePoint, mapping = mapping, data = data, stat = stat, position = position, show.legend = show.legend, inherit.aes = inherit.aes, params = list ( na.rm = na.rm, ...) ) } ggplot( mpg, aes( displ, hwy) ) + geom_simple_point( )
上面的代码和构建新的”stat”非常的相似,我们同样需要为4块内容提供属性/方法
required_aes: 用户所必需的提供的美术属性default_aes: 默认的图形属性值draw_key: 提供在图例(legend)绘制关键信息的函数,可用?draw_key查看帮助文档draw_panel: 这里就是见证奇迹的地方。该函数接受三个参数作为输入,返回一个grid的”grob”对象。它在每个面板(panel)运行一次。由于它是最复杂的内容,因此我们有必要详细地介绍它。
draw_panel有三个参数
data: 数据框,每一列都是一个图形属性panel_params: 一个列表,里面是coord产生的每个面板的参数。你需要将其当做一个不透明的数据结构: 不要看里面的细节,只要将其传递给coord方法。coord: 一个描述坐标系统的对象
你需要共同使用panel_params和coord才能对数据进行转换,即coords <- coord$transform(data, panel_params)。这会创建一个数据框,里面的位置变量会被缩放 到0-1之间。得到缩放数据用于调用”grid”的grob函数。(非笛卡尔坐标系统的数据转换比较复杂,你最好是将数据转成已有ggplot2的”geom”所接受的格式,然后传递)。
分组geoms 上一步我们用到的是draw_panel,也就是为每一行元素创建一个图形元素,比如说上面的GeomSimplePoint就是每一行一个点,这是最常见的情况。当然,如果你想为每一个分组绘制一个图形元素,那么我们应该使用draw_group()。
我们用一个简化版的GeomPolygon为例讲解这个知识点:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 GeomSimplePolygon <- ggproto( "GeomPolygon" , Geom, required_aes = c ( "x" , "y" ) , default_aes = aes( colour = NA , fill = "grey20" , size = 0.5 , linetype = 1 , alpha = 1 ) , draw_key = draw_key_polygon, draw_group = function ( data, panel_params, coord) { n <- nrow( data) if ( n <= 2 ) return ( grid:: nullGrob( ) ) coords <- coord$ transform( data, panel_params) first_row <- coords[ 1 , , drop = FALSE ] grid:: polygonGrob( coords$ x, coords$ y, default.units = "native" , gp = grid:: gpar( col = first_row$ colour, fill = scales:: alpha( first_row$ fill, first_row$ alpha) , lwd = first_row$ size * .pt, lty = first_row$ linetype ) ) } ) geom_simple_polygon <- function ( mapping = NULL , data = NULL , stat = "chull" , position = "identity" , na.rm = FALSE , show.legend = NA , inherit.aes = TRUE , ...) { layer( geom = GeomSimplePolygon, mapping = mapping, data = data, stat = stat, position = position, show.legend = show.legend, inherit.aes = inherit.aes, params = list ( na.rm = na.rm, ...) ) } ggplot( mpg, aes( displ, hwy) ) + geom_point( ) + geom_simple_polygon( aes( colour = class ) , fill = NA )
这里有几个注意点
我们重写了draw_group()而不是draw_panel(), 这是因为我们希望polygon是按照绘制,而不是按行绘制。
我们分组数据中不到两行,也就是没有足够的数据点去绘制polygon,因此我们返回了一个nullGrob()。你认为认为这是图形上的NULL: 这是一个grob对象,什么也不画,并且也不占任何空间
在单位上,x和y都应该是native的单位。(默认pointGrob()的单位就是native,因此我这里没有做修改)。多边形线的宽度(lwd)取决于点的大小,而ggplot2计算的点大小返回的mm 单位结果,因此作者将其和.pt相乘,将其调整为内部lwd接受的输入。
如果你将我们写的和实际的GeomPolygon比较,你会发现后者重写了draw_panel(),这是因为他用了一些小技巧来创建polygonGrob()从而在一次运行中得到多个polygon。这虽然更加复杂,但是在性能上更优秀。
从已有的Geom中继承 有些时候,你只想对已有的图层做一些小的修改。在这种情况下,除了从Geom继承以外,你还可以从已有的子类中继承。举个例子,我们可能想要更改GeomPolygon的默认值,使其更好的在StatChull中工作:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 GeomPolygonHollow <- ggproto( "GeomPolyHollwo" , GeomPolygon, default_aes = aes( colour = "black" , fill = NA , size = 0.5 , linetype = 1 , alpha = NA ) ) geom_chull <- function ( mapping = NULL , data = NULL , position = "identity" , na.rm = FALSE , show.legend = NA , inheirt.aes = TRUE , ...) { layer( stat = StatChull, geom = GeomPolygonHollow, data = data, mapping = mapping, position = position, show.legend = show.legend, inherit.aes = inheirt.aes, params = list ( na.rm = na.rm, ...) ) } ggplot( mpg, aes( displ, hwy) ) + geom_point( ) + geom_chull( )
尽管最终的geom_chull不允许你用更改”stat”对应的”geom”, 但是在当前的情况下,凸壳最应该用的”geom”应该就是多边形。
练习题
比较GeomPoint和GeomSimplePoint
比较GeomPolygon和GeomSimplePolygon
创建你自己的主题 如果你需要创建自己的完整主题,有以下几个件事情你需要知道
重写 已有的元素,而不是修改他们有四个 全局性元素影响几乎所有其他主题元素
完整和不完整元素的比较
重写元素 默认情况下,当你新增一个主题元素,它会从一个已有主题中继承参数值。例如,如下的代码设置key颜色是红色,但它继承了已有的fill颜色。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 theme_grey( ) $ legend.key new_theme <- theme_grey( ) + theme( legend.key = element_rect( colour = "red" ) ) new_theme$ legend.key
为了将其彻底重写,使用%+replace%而不是+:
1 2 3 4 5 6 7 8 9 new_theme <- theme_grey( ) %+replace% theme( legend.key = element_rect( colour = "red" ) ) new_theme$ legend.key
全局元素 有四个元素会影响绘图中的全局表现
Element
Theme function
Description
line
element_line()all line elements
rect
element_rect()all rectangular elements
text
element_text()all text
title
element_text()all text in title elements (plot, axes & legend)
很多特殊设置继承下来的属性都可以被以上这四个属性所修改。这对于修改整体背景颜色和总体字体非常有用。
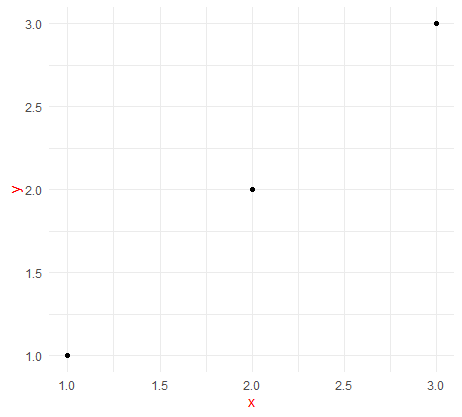
1 2 3 4 5 6 df <- data.frame( x = 1 : 3 , y = 1 : 3 ) base <- ggplot( df, aes( x, y) ) + geom_point( ) + theme_minimal( ) base
1 base + theme( text = element_text( colour = "red" ) )
建议在创建主题的起步阶段,先从修改这些值开始。
完整和不完整的比较 你需要理解完整主题对象 和不完整主题对象 之间的区别。一个完整的主题对象,就是一个主题函数中设置了complete=TRUE。
以theme_grey()和theme_bw()为例,他们就是完整的主题对象。而调用theme()则会得到一个不完整的主题对象。这两个区别在于,前者是对整体的修改,而后者只是修改了部分的元素。
1 2 3 4 attr ( theme_grey( ) , "complete" ) attr ( theme( ) , "complete" )
如果在一个完整对象上加上一个不完整对象,那么结果是一个完整对象
1 2 3 theme_test <- theme_grey( ) + theme( ) attr ( theme_test( ) , "complete" )
完整主题和不完整主题在添加到ggplot对象上有一些差别
在当前主题对象上增加一个不完整的主题对象,只会修改在theme()中定义的元素。
而在当前主题对象上增加一个完整主题对象,则会将已有主题完全覆盖成新的主题。
参考资料