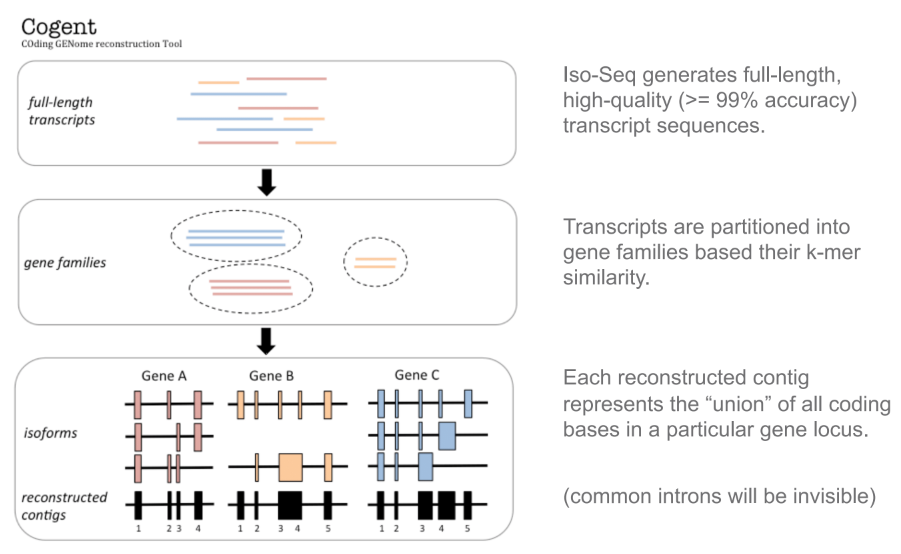
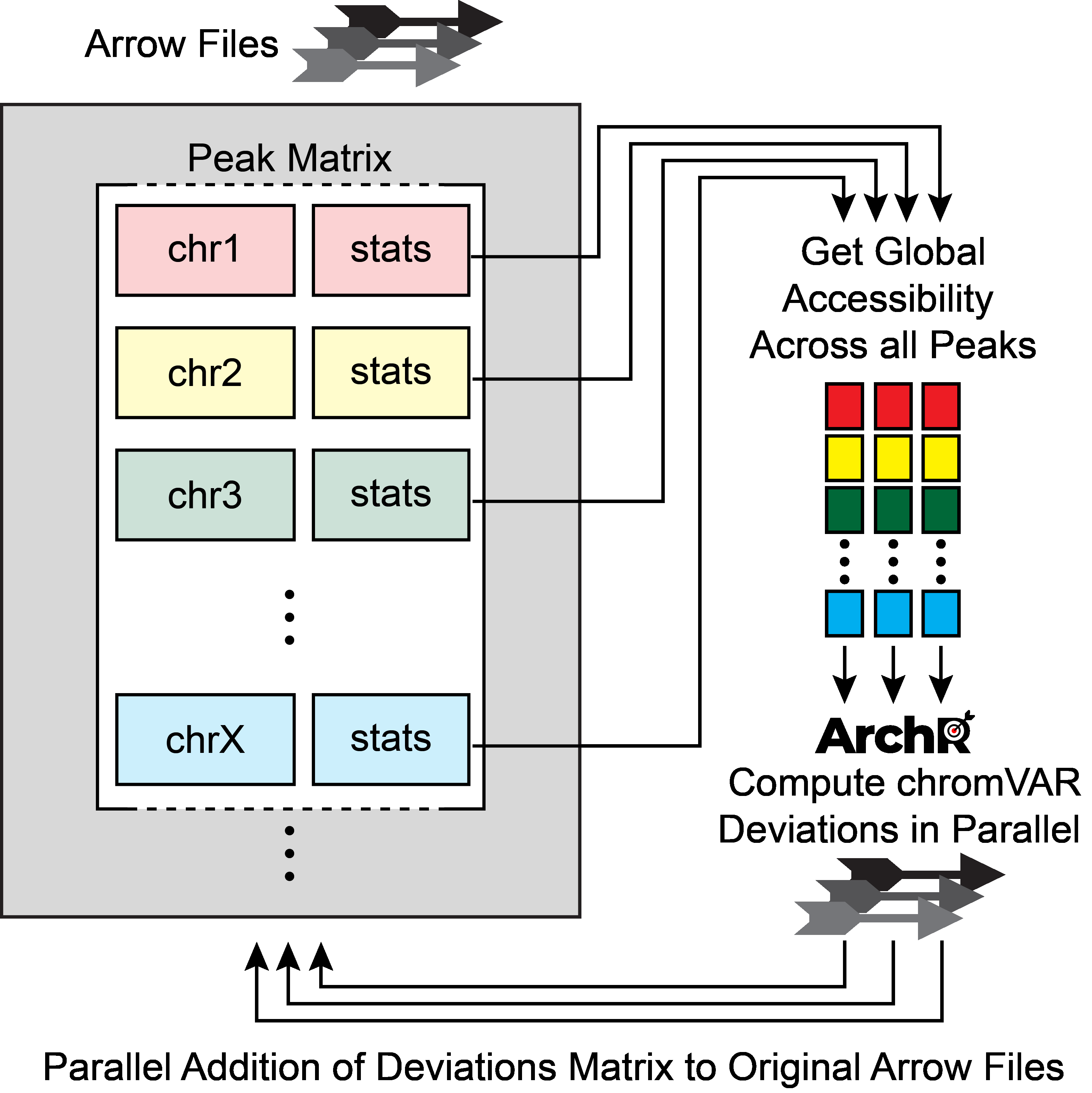
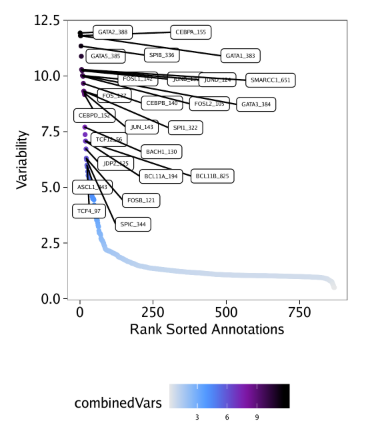
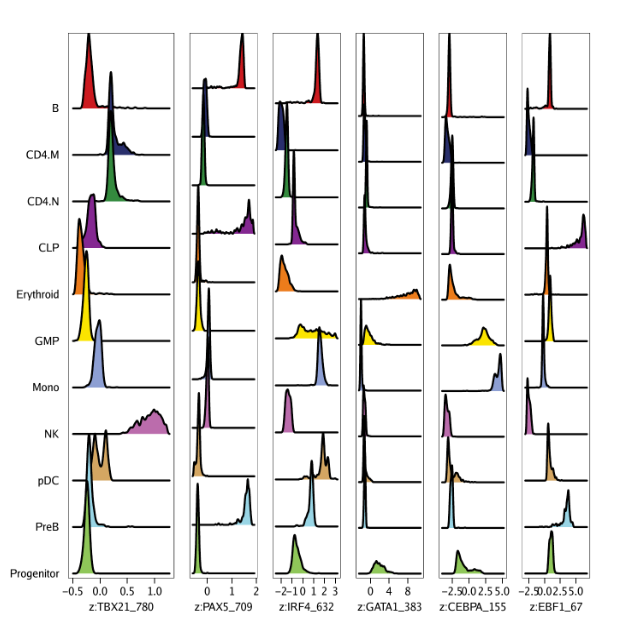
You do not need to be root (administrator on Windows) to install, use, or manage TeX Live. Indeed, we recommend installing it as a normal user, except perhaps on MacOSX, where it’s conventional to install as administrator.
======================> TeX Live installation procedure <=====================
======> Letters/digits in <angle brackets> indicate <======= ======> menu items for actions or customizations <=======
Detected platform: GNU/Linux on x86_64 <B> set binary platforms: 1 out of 6
<S> set installation scheme: scheme-full
<C> set installation collections: 40 collections out of 41, disk space required: 6516 MB
<D> set directories: TEXDIR (the main TeX directory): !! default location: /usr/local/texlive/2020 !! is not writable or not allowed, please select a different one! TEXMFLOCAL (directory for site-wide local files): /usr/local/texlive/texmf-local TEXMFSYSVAR (directory for variable and automatically generated data): /usr/local/texlive/2020/texmf-var TEXMFSYSCONFIG (directory forlocal config): /usr/local/texlive/2020/texmf-config TEXMFVAR (personal directory for variable and automatically generated data): ~/.texlive2020/texmf-var TEXMFCONFIG (personal directory forlocal config): ~/.texlive2020/texmf-config TEXMFHOME (directory for user-specific files): ~/texmf
<O> options: [ ] use letter size instead of A4 by default [X] allow execution of restricted list of programs via \write18 [X] create all format files [X] install macro/font doc tree [X] install macro/font source tree [ ] create symlinks to standard directories [X] after install, set CTAN as sourcefor package updates
<V> set up for portable installation
Actions: <I> start installation to hard disk <P> save installation profile to 'texlive.profile' and exit <H> help <Q> quit
Add /home/xzg/opt/texlive/texmf-dist/doc/man to MANPATH. Add /home/xzg/opt/texlive/texmf-dist/doc/info to INFOPATH. Most importantly, add /home/xzg/opt/texlive/bin/x86_64-linux to your PATH for current and future sessions. Logfile: /home/xzg/opt/texlive/install-tl.log
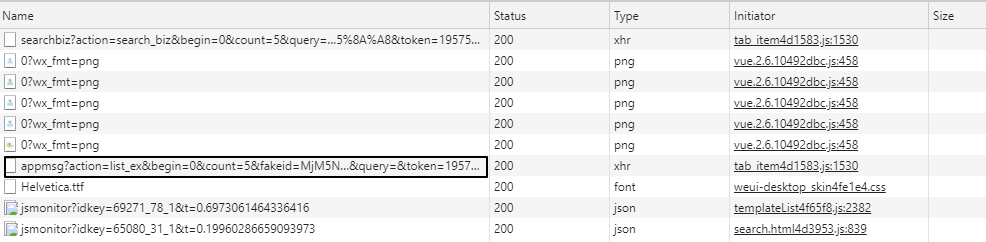
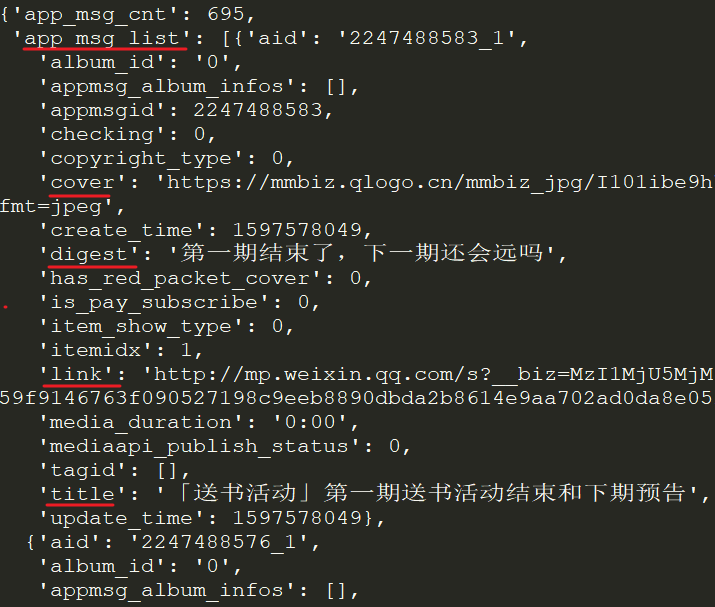
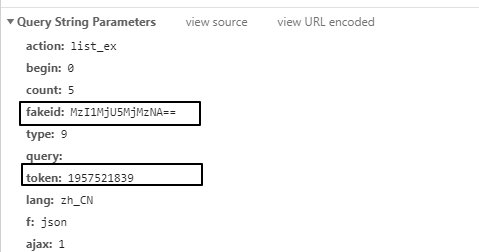
info_list = [] for msg in app_msg_list: if"app_msg_list"in msg: for item in msg["app_msg_list"]: info = '"{}","{}","{}","{}"'.format(str(item["aid"]), item['title'], item['link'], str(item['create_time'])) info_list.append(info) # save as csv withopen("app_msg_list.csv", "w") as file: file.writelines("\n".join(info_list))
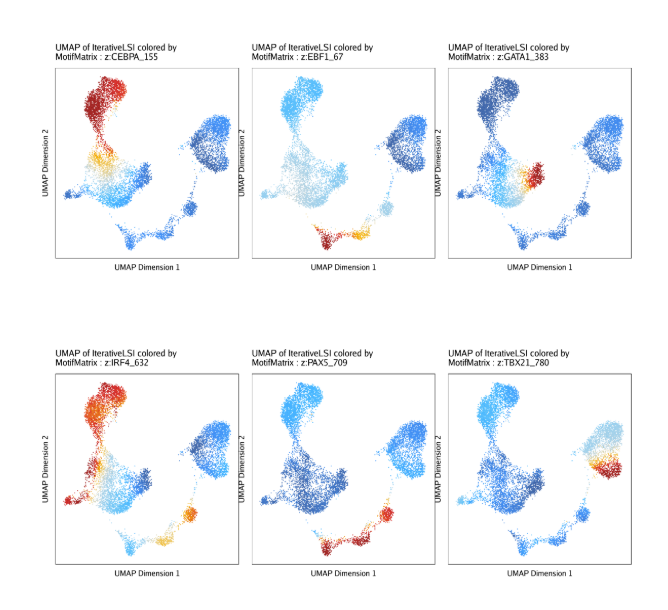
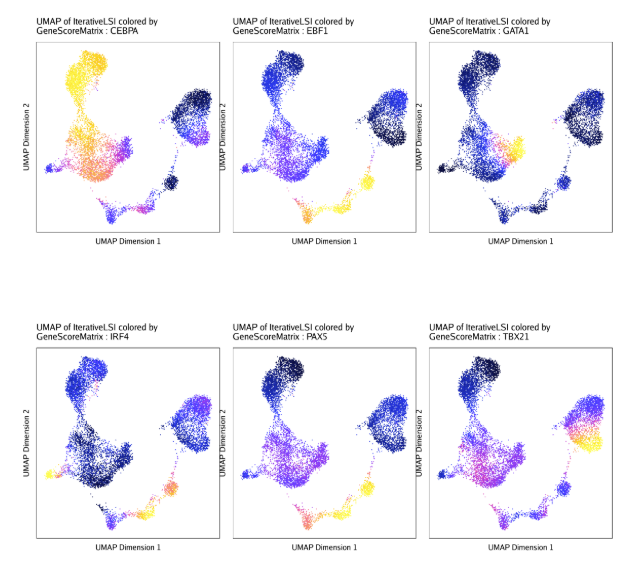
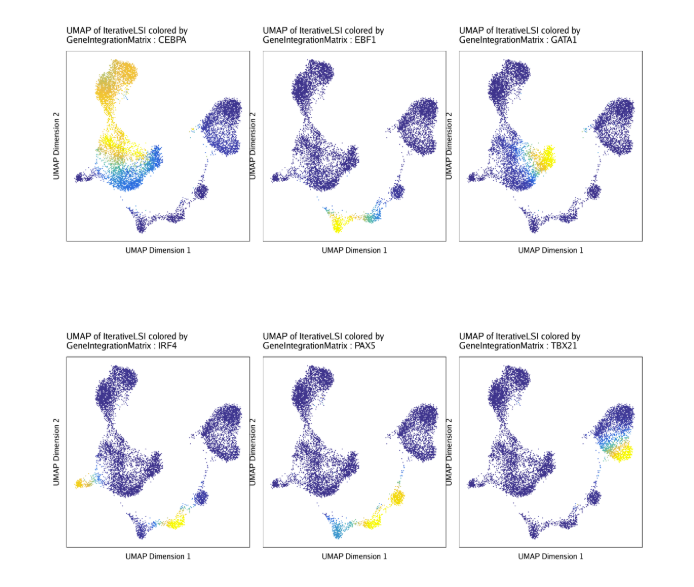
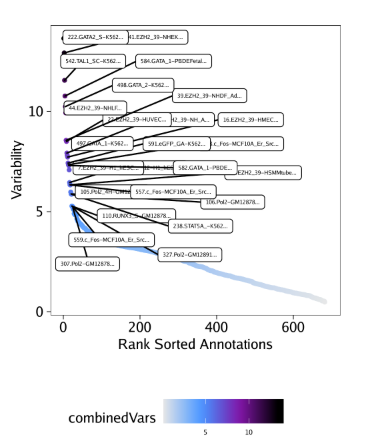
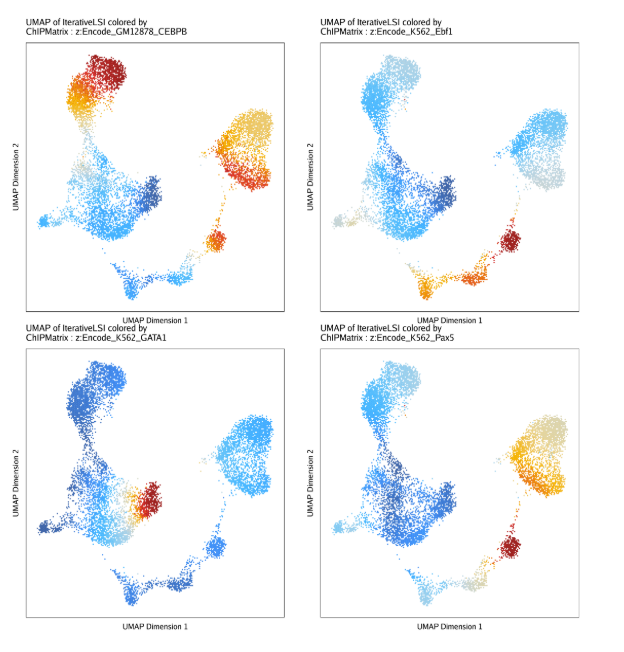
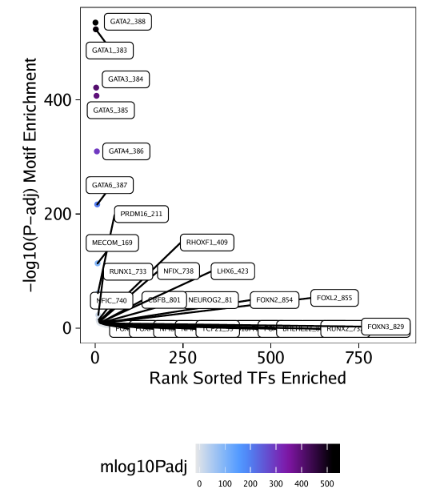
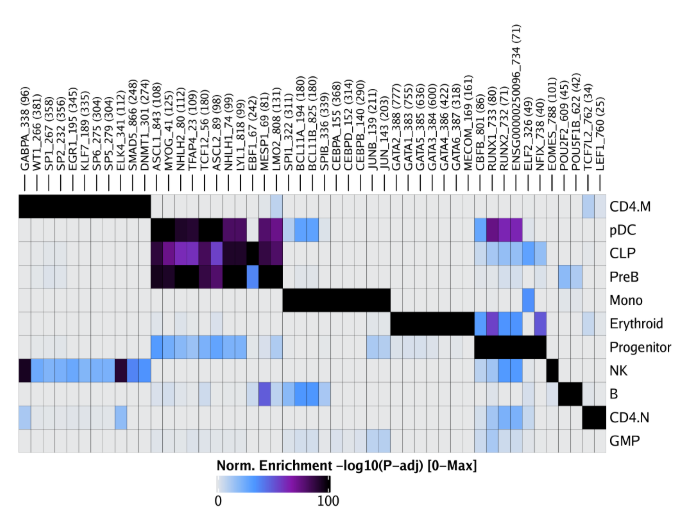
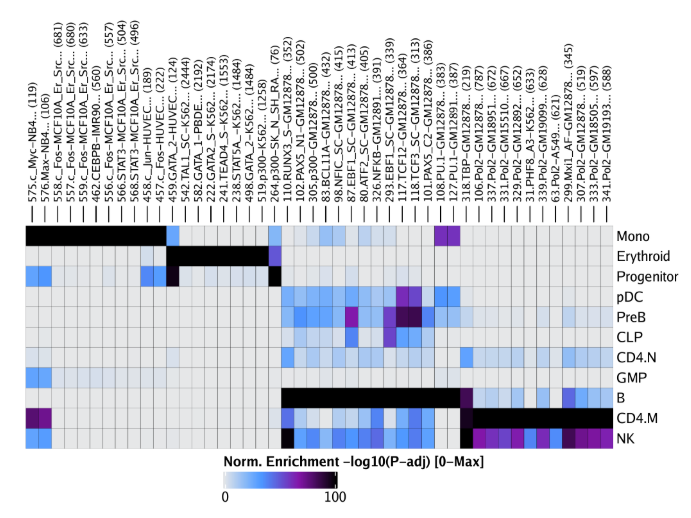
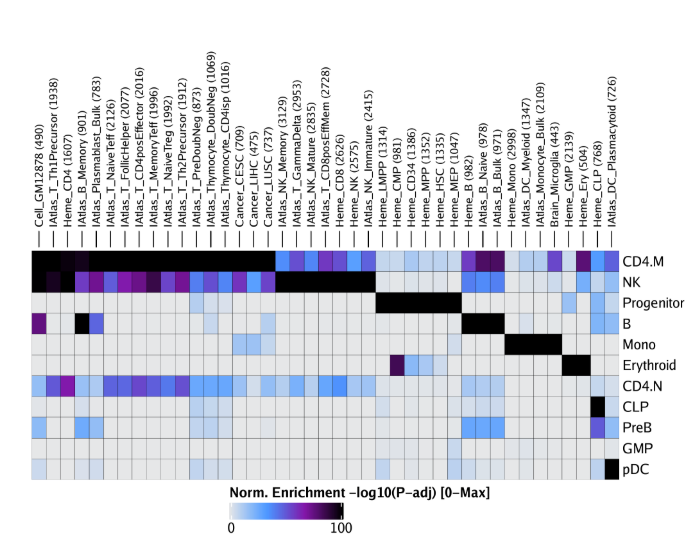
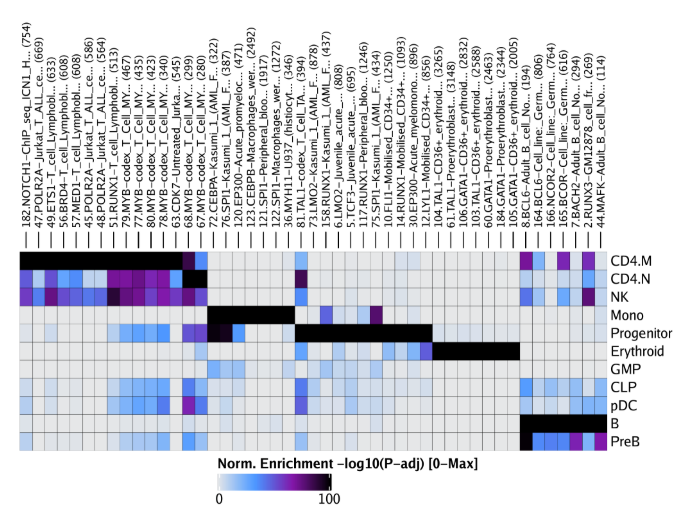
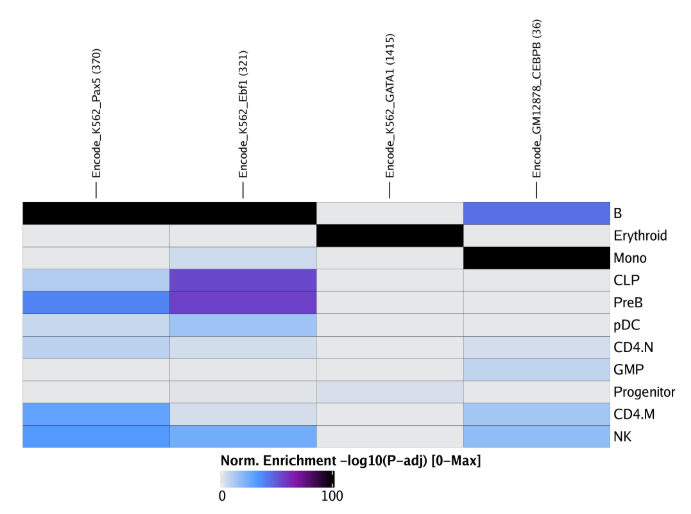
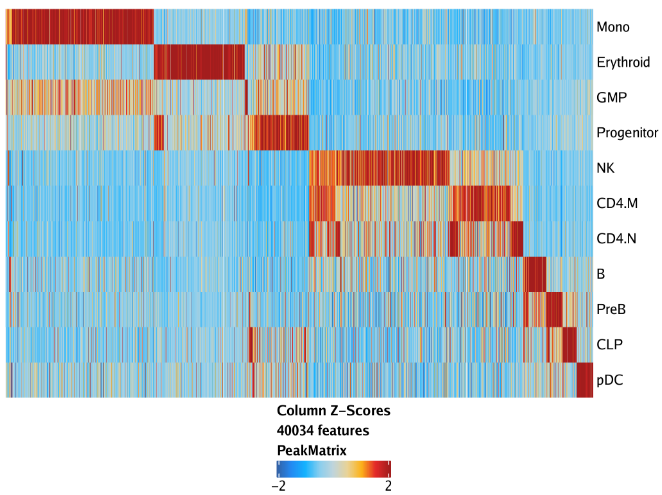
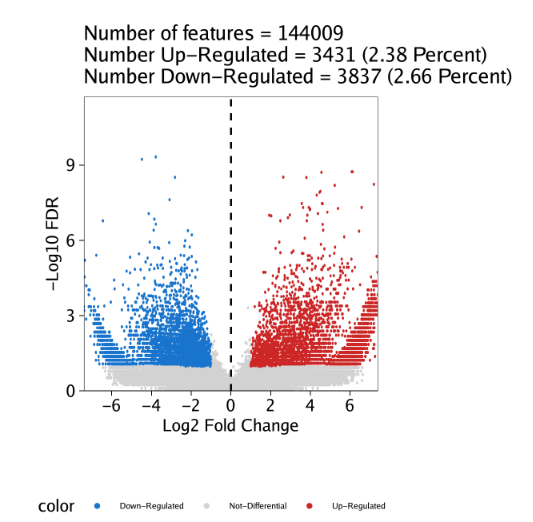
raw peak overlap就是分析不同细胞类型的peak是否有重叠,如果有将其合并成单个较大的peak。在如下的图解中,我们会发现尽管细胞类型A和C的前两个peak没有直接相连,但是由于细胞类型B中第一个peak和细胞类型A和C的前两个peak相互重叠,最终使得三个peak合并成一个更大peak。除了这个问题外,如果你想跟踪peak的顶点,你要么记录每一个合并后的新peak的顶点,要么记录每个合并后peak对应的原peak的顶点。
Searching For MACS2.. Not Found in$PATH Not Found with pip Not Found with pip3 Error in findMacs2() : Could Not Find Macs2! Please install w/ pip, add to your $PATH, or just supply the macs2 path directly and avoid this function!
#-----EST Evidence (for best results provide a file for at least one) est= #set of ESTs or assembled mRNA-seq in fasta format altest= #EST/cDNA sequence file in fasta format from an alternate organismest_gff= #aligned ESTs or mRNA-seq from an external GFF3 file altest_gff= #aligned ESTs from a closly relate species in GFF3 format
#-----Protein Homology Evidence (for best results provide a file for at least one) protein= #protein sequence file in fasta format (i.e. from mutiple organisms) protein_gff= #aligned protein homology evidence from an external GFF3 file
#-----Repeat Masking (leave values blank to skip repeat masking) model_org= #select a model organism for RepBase masking in RepeatMasker rmlib= #provide an organism specific repeat library in fasta format for RepeatMasker repeat_protein= #provide a fasta file of transposable element proteins for RepeatRunner rm_gff= #pre-identified repeat elements from an external GFF3 file prok_rm=0 #forces MAKER to repeatmask prokaryotes (no reason to change this), 1 = yes, 0 = no softmask=1 #use soft-masking rather than hard-masking in BLAST (i.e. seg and dust filtering)
#-----EST Evidence (for best results provide a file for at least one) est= #set of ESTs or assembled mRNA-seq in fasta format altest= #EST/cDNA sequence file in fasta format from an alternate organismest_gff=est.gff #aligned ESTs or mRNA-seq from an external GFF3 file est_gff=./est.gff altest_gff= #aligned ESTs from a closly relate species in GFF3 format
#-----Protein Homology Evidence (for best results provide a file for at least one) protein= #protein sequence file in fasta format (i.e. from mutiple organisms) protein_gff=protein2genome.gff #aligned protein homology evidence from an external GFF3 file
#-----Repeat Masking (leave values blank to skip repeat masking) model_org= #select a model organism for RepBase masking in RepeatMasker rmlib= #provide an organism specific repeat library in fasta format for RepeatMasker repeat_protein= #provide a fasta file of transposable element proteins for RepeatRunner rm_gff=repeats.gff #pre-identified repeat elements from an external GFF3 file prok_rm=0 #forces MAKER to repeatmask prokaryotes (no reason to change this), 1 = yes, 0 = no softmask=1 #use soft-masking rather than hard-masking in BLAST (i.e. seg and dust filtering)