准备阶段
训练SNAP模型,需要准备三个文件,分别是参考基因组序列,组装的转录本序列和同源蛋白序列。
对于参考基因组序列,我们要保证N50需要超过预期基因长度的中位数,否则注释效果不好。不过目前的基因组在三代测序的加持下,基本上都不是问题。
对于同源蛋白, 建议只用UniProt/Sprot的人工检查过的高质量蛋白序列,而不是盲目参考文献,使用近源物种的所有蛋白。除非你的近源物种都是模式物种,蛋白序列可靠性高,否则用错误的输入进行训练,数据越多反而错的越多。
我们可以在ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/taxonomic_divisions/选择合适的uniprot_sprot数据, 然后将其输出为fasta格式。以植物为例
1 | wget ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/taxonomic_divisions/uniprot_sprot_plants.dat.gz |
对于转录本,我们通常会测一些转录组数据,有三种策略可以得到转录本。(这里暂时不考虑三代全长转录本)
- Trinity重头组装转录本
- 使用STAR + Trinity 获取转录本
- 使用STAR + StringTie + gffread 获取转录本
对于这三种策略,不推荐策略一,因为在有参考基因组的情况下,策略二不但计算效率高,而且能避免组装错误(多倍体等位基因之间相似度高)。对于策略二和策略三,我会推荐策略三。因为对于靠的比较近的基因,Trinity很可能会把这两个基因装成一个。
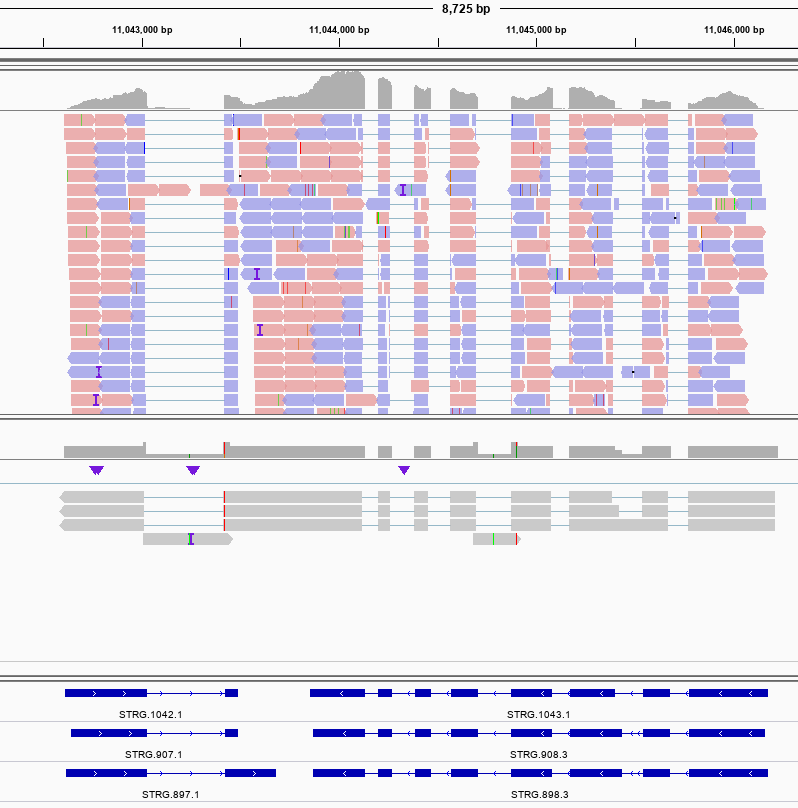
并且利用该转录本作为输入训练SNAP模型,之后以SNAP模型作为输入,将转录组和同源蛋白作为证据而不是直接用作模型,我们再检查maker的结果, 也会发现使用StringTie进行组装的结果才是对的。
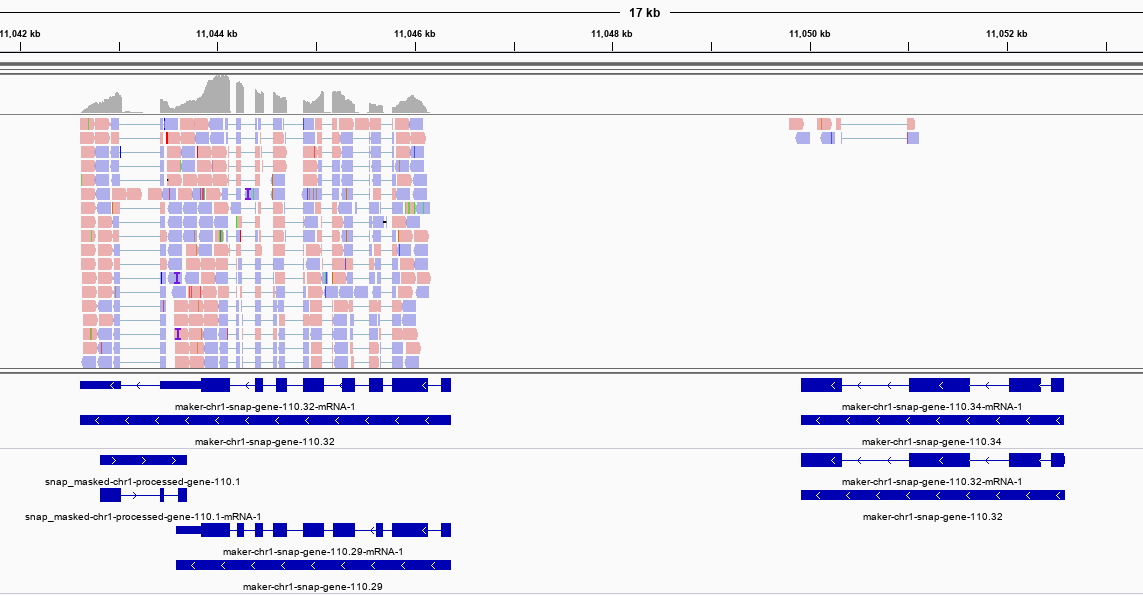
因此使用策略二不但计算量大,而且有些情况下还会导致过近的转录本错误融合,反而影响了最终效果,因此我最终推荐策略三。当然,这是我定性通过IGV浏览结果得出的结论,样本小,结论未必可靠,仅供参考。
训练阶段
假设我们准备的三个文件分别命名为, genome.fa, Trinity-GG.fasta 和 protein.fa
接着使用maker -CTL新建配置文件, 设置如下选项
1 | genome=genome.fa |
然后使用mpiexec -n 线程数 maker &> run.log运行程序。
处理结果后,我们新建一个snap目录训练模型
1 | mkdir snap && cd snap |
使用makerzff构建输入文件
1 | maker2zff -c 0.8 -e 0.8 -o 0.8 -x 0.2 genome.all.gff |
maker2zff会根据AED(-x)和QI值进行过滤,其中QI值一共有9项,每一项的含义如下
- Length of the 5’ UTR
- Fraction of splice sites confirmed by an EST alignment (
-c) - Fraction of exons that overlap an EST alignmetn(
-e) - Fraction of exons that overlap EST or Protein alignments(
-o) - Fraction of splice site confrimed by a ab-initio prediction(
-a) - Fraction of exons that overlap a ab-initio prediction(
-t) - Number of exons in the mRNA
- length of the 3’ UTR
- Length of the protein sequence produced by the mRNA (
-l)
如果QI值第二项为-1,表示没有支持该剪切位点的EST证据.
接着构建模型
1 | fathom -categorize 1000 genome.ann genome.dna |
修改配置,然后重新运行。MAKER会自动处理冲突的部分,避免重复序列屏蔽等的一些重复计算。
1 | genome=genome.fa |
根据输出结果再一次训练模型
1 | mkdir snap2 && cd snap2 |
通常迭代2-3次就够了,毕竟我们可能还会训练AUGUSTUS和GeneMark模型,通过比较多个模型来得到最终结果。