NextDenovo是武汉未来组胡江博士团队开发的一个三代组装工具,能够用于PacBio和Nanopore数据的组装。但是从官方的介绍而言,此工具在组装Nanopore上优势更大一些。
NextDenovo包括两个模块,NextCorrect用于原始数据纠错,NextGraph能够基于纠错后的进行组装。使用修改版的minimap2进行序列间相互比对。v2.0-beta.1版中在处理高度重复序列上可能存在错误组装,可以通过HiC和BioNano进行纠错。
软件安装
NextDenovo的软件安装非常简单, 下载解压缩即可使用。考虑到NextDenovo需要用Python2.7,我们可以用conda新建一个环境
1 | conda create -n python2 python=2.7 |
然后下载解压缩(我习惯把软件放在~/opt/bisofot下)
1 | mkdir -p ~/opt/biosoft/ |
测试下软件是否可以使用
1 | ~/opt/biosoft/NextDenovo/nextDenovo -h |
实战
以发表在NC上的拟南芥数据为例, 简单介绍下软件的使用
第一步: 新建一个分析项目
1 | mkdir NEXT && cd NEXT |
然后从EBI上下载该数据,在run.fofn中记录文件的实际位置。
1 | # 三代测序 |
第二步: 复制和修改配置文件
1 | cp ~/opt/biosoft/NextDenovo/doc/run.cfg . |
我的配置文件修改如下,参数说明参考官方文档
1 | [General] |
配置文件的几个重要参数说明(v2.0-beta.1)
- job_type 设置运行环境,可以使用(local, sge, pbs等)
- 运行线程数设置,线程数计算为
parallel_jobs分别与sort_option,minimap_options_*的-t数乘积,和correction_options的-p的乘积,量力而行。 - seed_cutfiles 如果在集群上运行,建议设置为可用的节点数,同时设置
correction_options的-p为各个节点可用的核数,保证每个节点只有一个correction任务,减少运行时的内存和IO。 如果local上运行, 建议设置为总可用的核除以correction_options的-p值. - parallel_jobs建议设置至少要大于
pa_correction。 - blocksize 是将小于seed_cutfiles的数据拆分成的多个文件时单个文件的大小, 总的比对任务数等于基于该参数切分的文件数乘以
seed_cutfiles + seed_cutfiles * (seed_cutfiles - 1)/2, 因此对于10g以内的数据量, 建议设置小于1g, 避免总的任务数小于parallel_jobs的值。 - 测序数据类型相关: 对于PacBio而言,要修改
minimap2_options_*中的-x ava-ont为-x ava-pb - 数据量相关参数:
read_cutoff = 1k过滤原始数据中低于1k的read,seed_cutoff = 30k则是选择大于30k以上的数据来矫正。关于seed_cutoff的设置,可以通过~/opt/biosoft/NextDenovo/bin/seq_stat来获取参考值,不建议直接使用默认值,因为改值会受到测序深度和测序长度影响,而且一个不合适的值会显著降低组装质量。对于基因组大于200m以上的物种,-d建议默认。 - correction_options中的-dbuf可以显著降低矫正时的内存,但会显著降低矫正速度。
- random_round参数,建议设置20-100. 该参数是设置随机组装参数的数量,nextGraph会基于每一套随机参数做一次组装, 避免默认参数效果不好。
seq_stat能够根据物种大小和预期用于组装的深度确定seed_cutoff
1 | ~/opt/biosoft/NextDenovo/bin/seq_stat -g 110Mb -d 30 input.fofn |
第三步: 运行NextDenovo
1 | ~/opt/biosoft/NextDenovo/nextDenovo run.cfg & |
运行时间如下
1 | real 64m5.356s |
默认参数结果是存放在01_rundir/03.ctg_graph/01.ctg_graph.sh.work/ctg_graph00, 可以将其复制到当前目录,用于后续的分析。
1 | cat 01_rundir/03.ctg_graph/01.ctg_graph.sh.work/ctg_graph00/nextgraph.assembly.contig.fasta > nextgraph.assembly.contig.fasta |
但是在01.ctg_graph.sh.work目录下除了ctg_graph00以外,还有其他随机参数的在组装结果。随机参数结果只输出了统计结果,用户如需要输出组装序列,可以修改01_rundir/03.ctg_graph/01.ctg_graph.sh,将里面的-a 0替换成-a 1。
每个目录下都有shell输出,可以挑选基于nextDenovo.sh.e这里面的结果挑选组装指标较好的,再输出序列,比如说比较下N50
1 | grep N50 01_rundir/03.ctg_graph/01.ctg_graph.sh.work/ctg_graph*/*.e |
默认情况下,最终组装出20条contig,总大小116M,N50 12M.
使用minimap2将组装结果和比对到TAIR10上,用dotplotly进行可视化
1 | minimap2 -t 100 -x asm5 Athaliana.fa nextgraph.assembly.contig.fasta > next.paf |
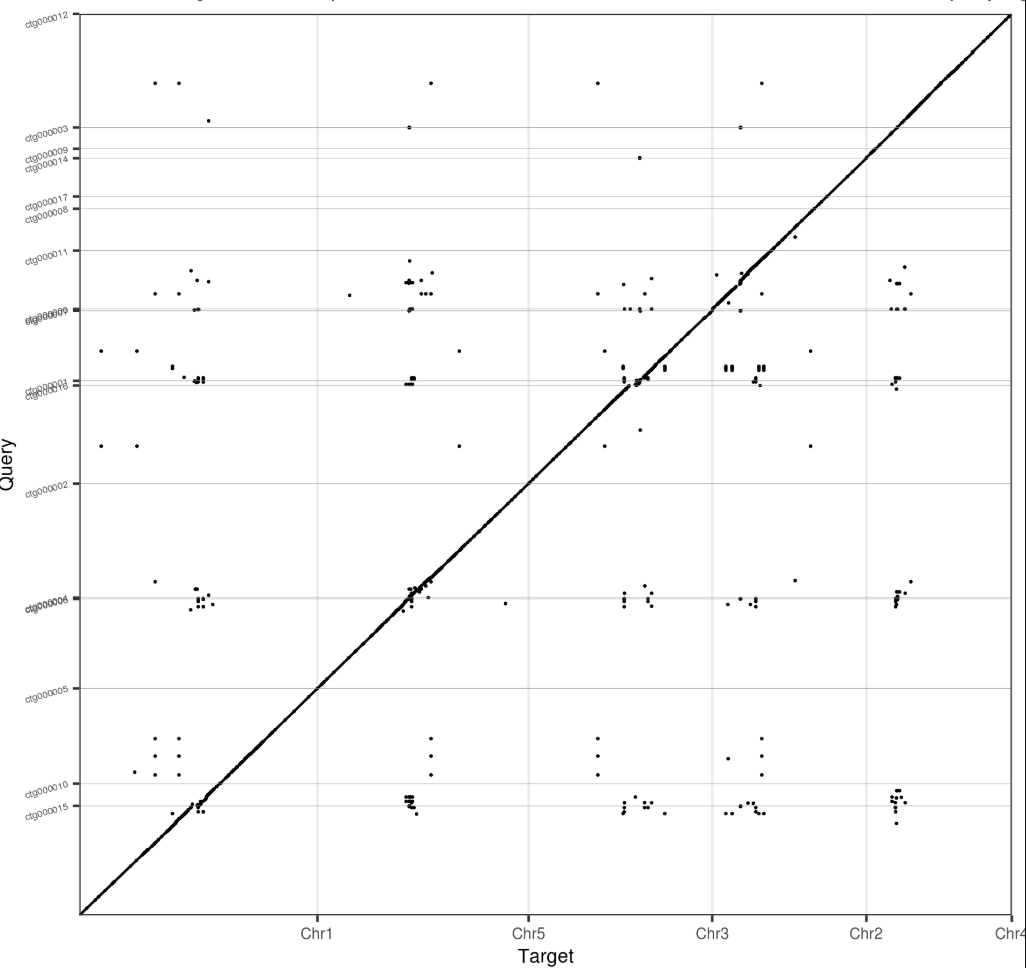
不难发现,两者存在高度的共线性。大部分TAIR10的染色体对应的都是2条或者3条contig。
此外这篇NC的拟南芥提供了BioNano光学图谱,我使用BioNano Hyrbrid Scaffold 流程进行了混合组装
1 | cp /opt/biosoft/Solve3.3_10252018/HybridScaffold/10252018/hybridScaffold_config.xml . |
组装结果如下,从原来的20的contig下降到了16个contig。
1 | Count = 16 |
此外还通过BioNano Access进行可视化,以其中一个结果为例。光学图谱和NextDenovo的组装结果存在很高的一致性。
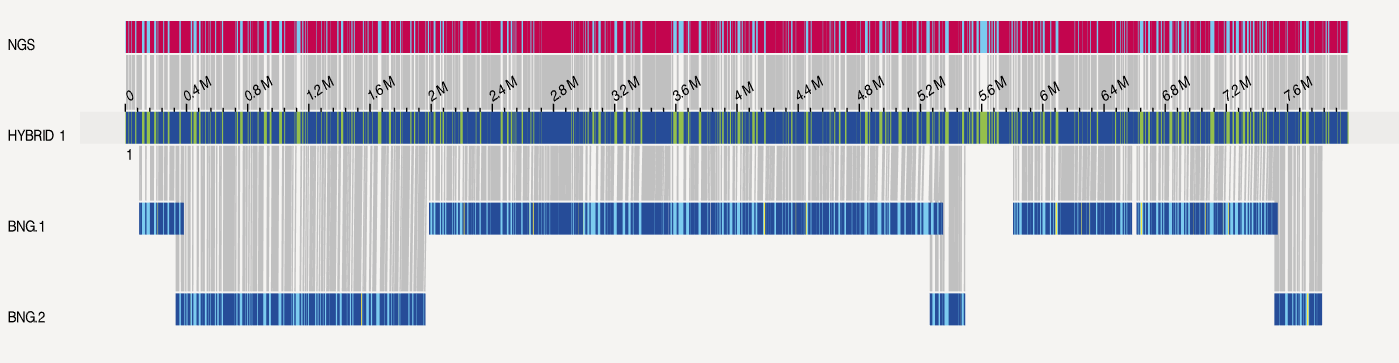
综上,在Nanopore上组装上,我们又多了一个比较好用的工具。
最后非常感谢胡江博士对于本文的指导!