软件安装
软件的GitHub地址为https://github.com/alexdobin/STAR, 下载页面为https://github.com/alexdobin/STAR/releases, 挑最新的下载,避免bug。
软件使用
STAR的主程序只有两个:STAR和STARlong。前者用于比对RNA-seq数据,后者是针对于长读长RNA数据。由于同一个程序,又需要做建索引,又需要做序列比对,并且这个程序还支持一系列的输出格式,因此直接用STAR,你会迷失在参数的海洋中。所以我们需要先阅读文档 ,先对整体有一个了了解。
STAR的基本使用流程分为两步:
- 生成基因组索引文件。你需要提供基因组序列(FASTA)和注释文件(GTF)
- 将读段回帖到基因组。这一步需要提供的是RNA-seq数据,格式目前都是FASTQ/FASTA, 最后会得到很多很多的文件,常规的SAM/BAM文件,可变剪切点文件,未回帖上的读段和常用于展示信号的WIG文件。
STAR的使用格式为
1 | STAR --option1-name option1-value --option2-name option2-value ... |
建立索引
举例说明:
1 | STAR --runMode genomeGenerate \ |
常用参数说明
- –runThreadN 线程数 :设置线程数
- –runMode genomeGenerate : 设置模式为构建索引
- –genomedDir 索引文件存放路径 : 必须先创建文件夹
- –genomeFastaFiles 基因组fasta文件路径 : 支持多文件路径
- –sjdbGTFfile gtf文件路径 : 可选项,高度推荐,用于提高比对精确性
- –sjdbOverhang 读段长度: 后续回帖读段的长度, 如果读长是PE 100, 则该值设为100-1=99
几个额外要说明的点:
- 由于物种的组装的复杂性,存在一些为组装上的片段,这些片段不需要放在参考序列中,尤其是可变单倍型(alternative haplotypes)
- 如果基因组的contig过多,超过5000,你需要用
--genomeChrBinNbits=min(18,log2[max(GenomeLength/NumberOfReferences,ReadLength)])降低RAM消耗 - 选择最新的注释文件,人类和小鼠常在http://www.gencodegenes.org下载,植物的可信基因组见http://plants.ensembl.org
- 如果没有设置
--sjdbGTFfile或--sjdbFileChrStartEnd,就不需要设置--sjdbOverhang
读段回帖
用法举例
1 | STAR \ |
参数说明
- –runThreadN 设置线程数
- –runMode alignReads : 默认就是比对模式,可以不填写
- –genomeDir: 索引文件夹
- –readFilesIn FASTA/Q文件路径
- –readFilesCommand zcat: 如果输入格式是gz结尾,那么需要加上zcat, 否则会报错
- –outSAMtype: 输出SAM文件的格式,是否排序
- –outBAMsortingThreadN: SAM排序成BAM时调用线程数
默认参数下,会输出文件在当前文件夹,
1 | Aligned.out.sam Log.final.out Log.out Log.progress.out SJ.out.tab |
可以用--outFileNamePrefix指定文件夹和前缀,其中”Aligned.out.sam”是默认回帖后输出。一般而言,SAM文件过大,不方便后续使用,我们更需要的是BAM文件。最好是类似于samtools sort的输出文件,那么设置参数为--outSAMtype BAM SortedByCoordinate。 如果你设置的线程数非常大,那么你很有可能会遇到如下这种报错,我的解决方案就是降低线程数。
1 | FATAL ERROR: number of bytes expected from the BAM bin does not agree with the actual size on disk |
xxx.out文件是一些日志信息.
“SJ.out.tab”存放的高可信的剪切位点,每一列的含义如下
- 第一列: 染色体
- 第二列: 内含子起始(以1为基)
- 第三列: 内含子结束(以1为基)
- 第四列:所在链,1(+),2(-)
- 第五列: 内含子类型,0表示不是下面的任何一种功能,1表示GT/AG, 2表示:GT/AC,3表示GC/AG,4表示GT/GC,5表示AT/GC,6表示GT/AT
- 第六列: 是否是已知的注释
- 第七列: 有多少唯一联配支持
- 第八列: 有多少多重联配支持
- 第九列: maximum spliced alignment overhang, 这个比较难以翻译,指的是当短读比对到剪切位点时,中间会被分开,另一边能和基因组匹配的数目,例如ACGTACGT———-ACGT,就是4或者8,取决于方向。
控制过滤的参数为--outSJfilter*系列,其中--outSJfilterCountUniqueMin 3 1 1 1表示4类内含子唯一匹配的read支持数至少为3,1,1,1, 而--outSJfilterCountTotalMin 3 1 1 1则表示4类内含子唯一匹配和多重匹配read的支持数和,至少为3,1,1,1。如果你设置的--outSJfilterReads Unique,那么上面两者是等价的,当然默认情况下是All
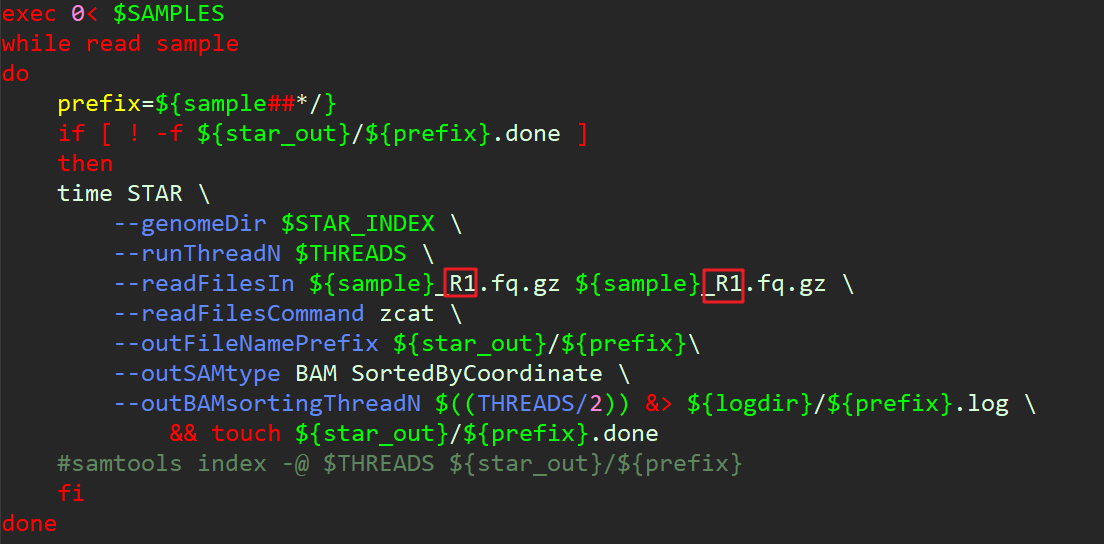
注意:双端测序中一定要注意不能把文件输入错了,不然比对率几乎为0。下面我一次脚本翻车记录,两个都是R1和R1,应该是R1和R2
更多参数
除了上面常用的一些参数外,STAR的可选参数其实非常多.
输出BAM文件时,STAR还可以对BAM进行一些预处理,”–bamRemoveDuplicatesType”用于去重(“UniqueIdentical”,”UniqueIdenticalNotMulti”)
如果你希望输出信号文件(Wig格式),那么需要额外增加--outWigType参数,如--outWigType wiggle read2, 还可以用--outWigStrand指定是否将两条链合并(Stranded, Unstranded), 默认--outWigNorm RPM,也就是用RPM进行标准化,可以选择None.
如果你在建立索引或者比对的时候增加了注释信息,那么STAR还能帮你进行基因计数。参数为--quantMode, 分为转录本水平(TranscriptomeSAM)和基因水平(GeneCounts),在计数的时候还允许指定哪些哪些read不参与计数,”IndelSoftclipSingleend”和”Singleend”
对于非链特异性RNA-seq,同时为了保证能和Cufflinks兼容,需要添加--outSAMstrandField intronMotif在SAM中增加XS属性,并且建议加上--outFilterIntronMotifs RemoveNoncanonical。如果是链特异性数据,那么就不需要特别的参数,Cufflinks用--library-type声明类型即可=