这一篇会在之前的基础上开始在circos绘制基因密度信息。为了保证一致性,可以新建如下几个文件
circos.conf:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 karyotype = karyotype.tair10.txt chromosomes_color = chr1=rdylbu-11-div-1,chr2=rdylbu-11-div-3,chr3=rdylbu-11-div-5,chr4=rdylbu-11-div-7,chr5=rdylbu-11-div-9 chromosomes_units = 1000000 <<include ticks.conf>> <ideogram> <spacing> default = 0.005r </spacing> radius = 0.90r thickness = 20p fill = yes stroke_color = dgrey stroke_thickness = 2p show_label = yes #展示label label_font = default # 字体 label_radius = dims(ideogram,radius) + 0.05r #位置 label_size = 16 # 字体大小 label_parallel = yes # 是否平行 label_format = eval(sprintf("%s",var(chr))) # 格式 </ideogram> <image> dir* = . # 输出文件夹 radius* = 500p # 图片半径 svg* = no # 是否输出svg <<include etc/image.conf>></image> <<include etc/colors_fonts_patterns.conf>> <<include etc/housekeeping.conf>>
karyotype.tair10.txt
1 2 3 4 5 chr - chr1 chr1 0 30427617 chr1 chr - chr2 chr2 0 19698289 chr2 chr - chr3 chr3 0 23459830 chr3 chr - chr4 chr4 0 18585056 chr4 chr - chr5 chr5 0 26975502 chr5
ticks.conf
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 show_ticks = yes show_tick_labels = yes <ticks> radius = 1r color = black thickness = 2p multiplier = 1e-6 format = %d <tick> spacing = 1u size = 5p </tick> <tick> thickness = 4p spacing = 5u size = 10p show_label = yes label_size = 10p label_offset = 10p format = %d </tick> </ticks>
在开始之前,请确保已经安装了bedtools,如果没有的话,用conda安装
1 2 conda install -c bioconda bedtools
数据格式 为了能够在circos绘制基因密度信息,需要先知道circos要求的输入数据格式是什么。
对于折线图(line),散点图(scatter),柱状图(histogram)和热图(heatmap),Circos要求的数据输入格式相同,也就是chr start end value [options], 如果熟悉BED格式定义的话,你就会发现除了可选(options)外,Circos要求的格式就是4列的BED。
对于可选列,可以和circos.conf里的<rule>搭配使用,属于比较高级的用法。
数据预处理 为了能够获得所需的基因密度信息,我们需要下载拟南芥的GFF文件。
1 2 wget ftp://ftp.ensemblgenomes.org/pub/plants/release-44/gff3/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.44.gff3.gz
提取基因的位置信息
1 zgrep '[[:blank:]]gene[[:blank:]]' Arabidopsis_thaliana.TAIR10.44.gff3.gz | cut -f 1,4,5 | awk '{print "chr"$1"\t"$2"\t"$3}' > genes.bed
接着用bedtools以500kb为滑窗,沿染色体创建窗口
1 2 cut -d ' ' -f 3,6 karyotype.tair10.txt | tr ' ' '\t' > tair10.genomebedtools makewindows -g tair10.genome -w 500000 > tair10.windows
最后统计信息
1 bedtools coverage -a tair10.windows -b genes.bed | cut -f 1-4 > genes_num.txt
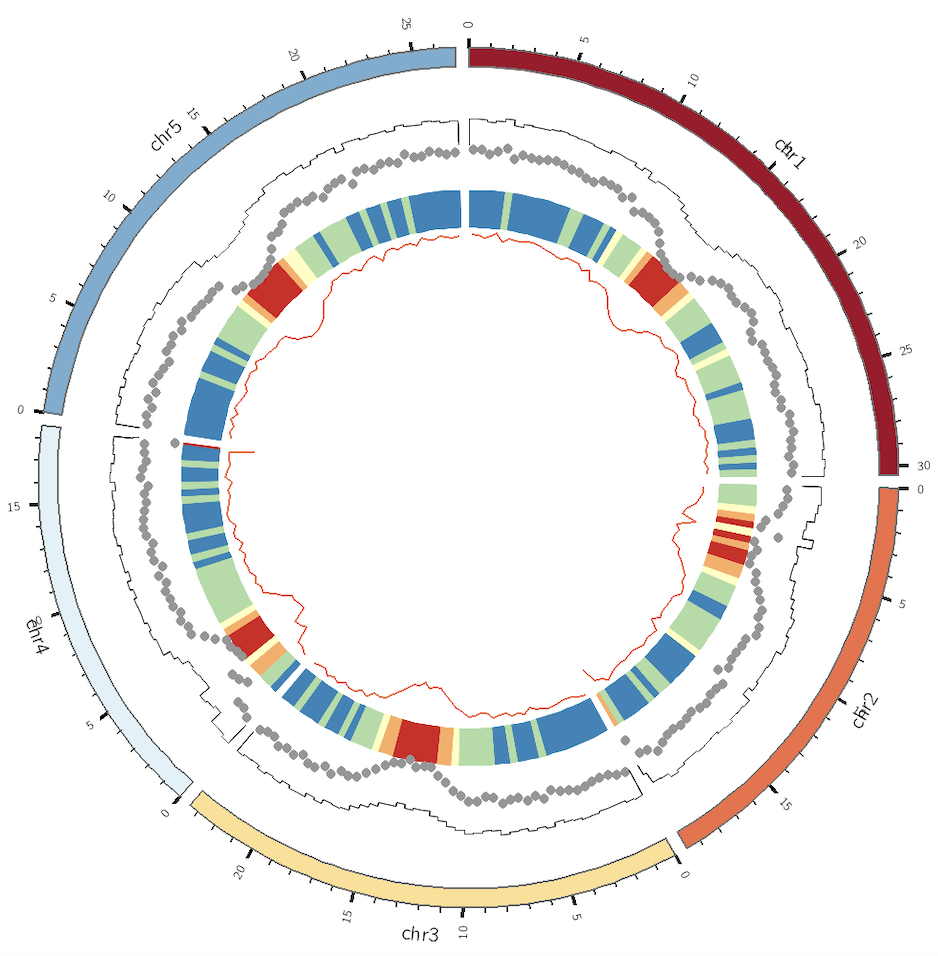
展示信息 我们可以先用最少的参数,同时展示不同的图形。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 ... <plots> <plot> type = line thickness = 2 max_gap = 1u file = genes_num.txt color = redv r0 = 0.51r r1 = 0.60r </plot> <plot> type = heatmap file = genes_num.txt color = spectral-5-div r1 = 0.70r r0 = 0.61r </plot> <plot> type = scatter fill_color = grey stroke_color = black glyph = circle glyph_size = 10 file = genes_num.txt r1 = 0.80r r0 = 0.71r </plot> <plot> type = histogram file = genes_num.txt r1 = 0.89r r0 = 0.81r </plot> </plots> ...
运行之后,效果如下
这张图展示了巨大的进步空间,至少可以从如下几个角度进行调整,
柱状图有点丑,需要填充颜色
热图的颜色不符合直觉,基因数越少反而颜色越深
散点图的点太大
有些图形可能需要加上背景色。
对于一些过大的数值,最好用一种颜色表示。
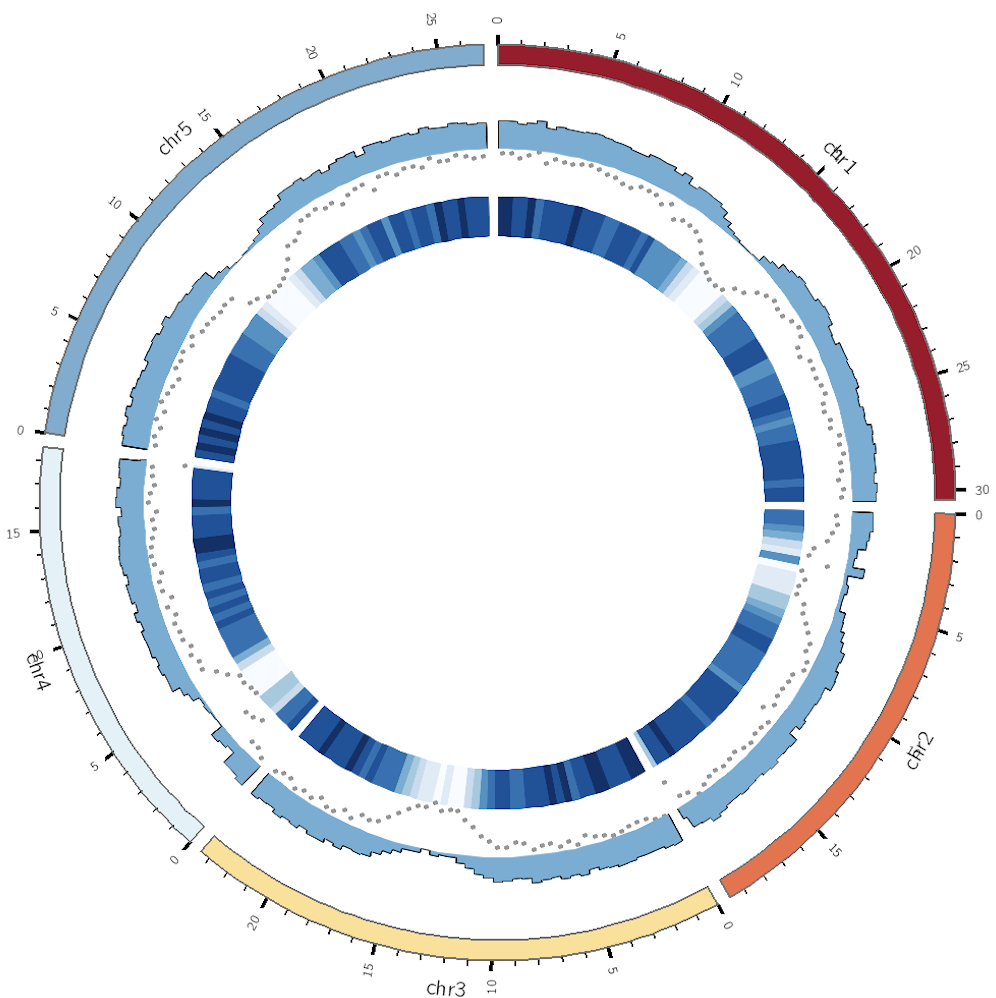
当知道自己的目标后,后续的事情就是找对应参数和调整参数. 部分我用来调整参数如下
show - 是否展示该图形type - 展示的图形类型file - 输入的数据文件所在路径min/max - 数据范围r0/r1 - 内径和外径,在圈图中的位置glyph - 对于散点图 而言,还可以选择符号的类型,是circle, rectangle, 还是 triangleglyph_size - 符号的大小,单位为pcolor 散点图符号颜色,柱状图外部线的颜色stroke_color - 对于散点图,符号外部是否也要颜色stroke_thickness - 对于散点图,符号外部线的厚度fill_color:柱状图填充色
调整后的参数为
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 ... <plots> <plot> type = heatmapfile = genes_num.txt color = blues-9-seq r1 = 0.70r r0 = 0.61r </plot> <plot> type = scatterfill_color = black stroke_color = black glyph = circle glyph_size = 5 file = genes_num.txt r1 = 0.80r r0 = 0.71r </plot> <plot> type = histogramfile = genes_num.txt fill_color = blue r1 = 0.89r r0 = 0.81r </plot> </plots> ...
图形效果如下
这个效果我个人还是比较满意的。更加复杂的设置,目前还不适合我,需要一步一步来。
下一部分将会介绍如何在circos展示两个物种的共线性,也就是circos不同染色体之间的连线。