from transformers import pipeline generator = pipeline(task="text-generation", model=model, tokenizer=tokenizer)
# 英文输入 generator("bioinformatics is ", max_length=30, num_return_sequence=5)
# 输出结果 [{'generated_text': 'bioinformatics is a branch of computer science that deals with the analysis of data and the design of algorithms that can be used to solve problems in'}]
> png('ab.png') Error in .External2(C_X11, paste0("png::", filename), g$width, g$height,: unable to start device PNG In addition: Warning message: In png("ab.png"): unable to open connection to X11 display ''
$ sudo lsof /Volumes/Time-Machine COMMAND PID USER FD TYPE DEVICE SIZE/OFF NODE NAME mds 334 root 27r DIR 1,29 160 2 /Volumes/Time-Machine
结果显示,有一个PID为334的mds的进程正在使用硬盘。那么问题来了,这个mds是啥呢? 使用百度加上 man mds, 我发现这是MacOS上的一个索引工具,同时我还找到一位跟我有类似经历的文章。简单说,就是因为时间机器备份需要进行文件的比对,为了实现这种快速的文件件对比,那么我们就需要建立文件索引。
#案例代码 deftest_arguments(first, *middle, **last): print(f"first is: {first}") for i,j inenumerate(middle): print(f"the {i} of middle is {j}") for k,v in last.items(): print(f"key {k} is value {v}")
In order to test for differential expression, we operate on raw counts and use discrete distributions as described in the previous section on differential expression. However for other downstream analyses – e.g. for visualization or clustering – it might be useful to work with transformed versions of the count data. Analyzing RNA-seq data with DESeq2
To identify correlated cell types between two cell atlas datasets, we first aggregated the cell-type-specific UMI counts, normalized by the total count, multiplied by 100,000 and log-transformed after adding a pseudocount
To improve accuracy and specificity, we selected cell-type-specific genes for each target cell type by (1) ranking genes on the basis of the expression fold-change between the target cell type versus the median expression across all cell types, and then selecting the top 200 genes; (2) ranking genes on the basis of the expression fold-change between the target cell type versus the cell type with maximum expression among all other cell types, and then selecting the top 200 genes; and (3) merging the gene lists from steps (1) and (2).
Similarly, we then switch the order of datasets A and B, and predict the gene expression of target cell type (Tb) in dataset B with the gene expression of all cell types (Ma) in dataset A.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
# predict a with b seu.obj.gene <- seu.obj.mat2[, cluster]
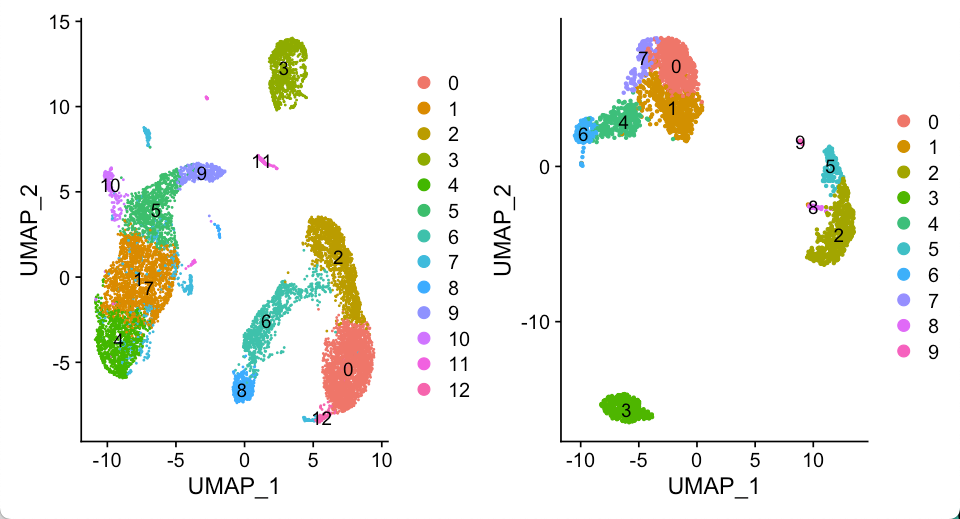
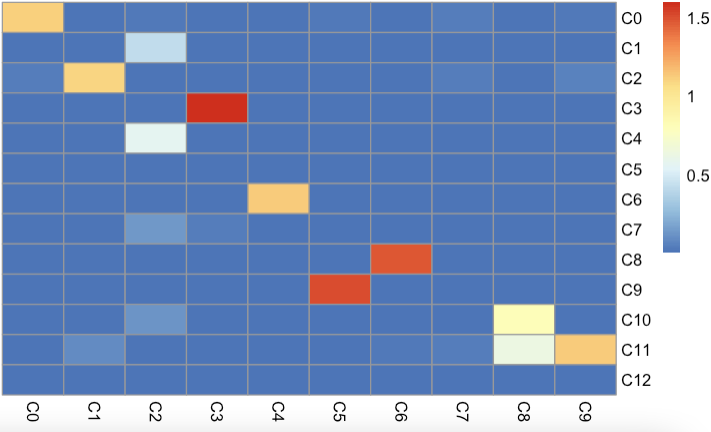
For validation, we first applied cell-type correlation analysis to independently generated and annotated analyses of the adult mouse kidney (sci-RNA-seq component of sci-CAR19 versus Microwell-seq). We subsequently compared cell subclusters from this study (with detected doublet-cell ratio ≤10%) to fetus-related cell types (those with annotations including the term ‘fetus’) from the Microwell-seq-based MCA. A similar comparison was performed against cell types annotated in BCA.