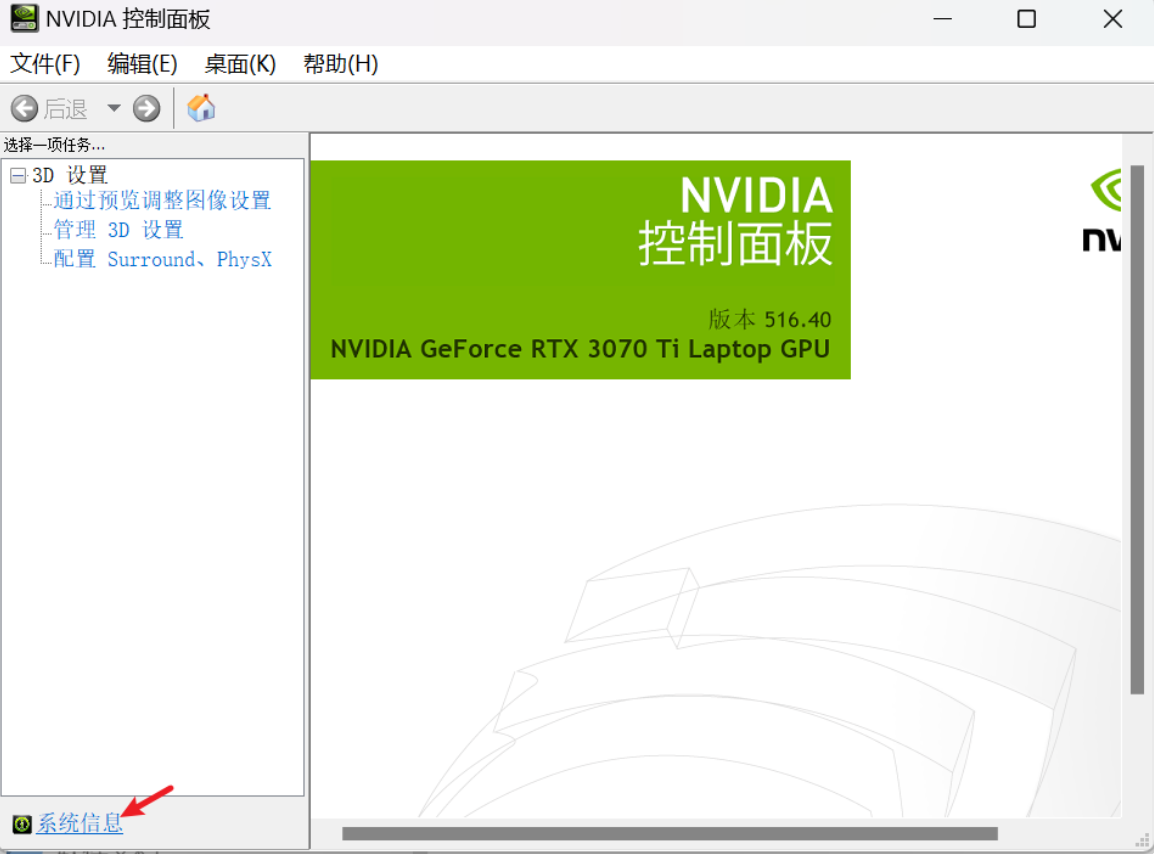
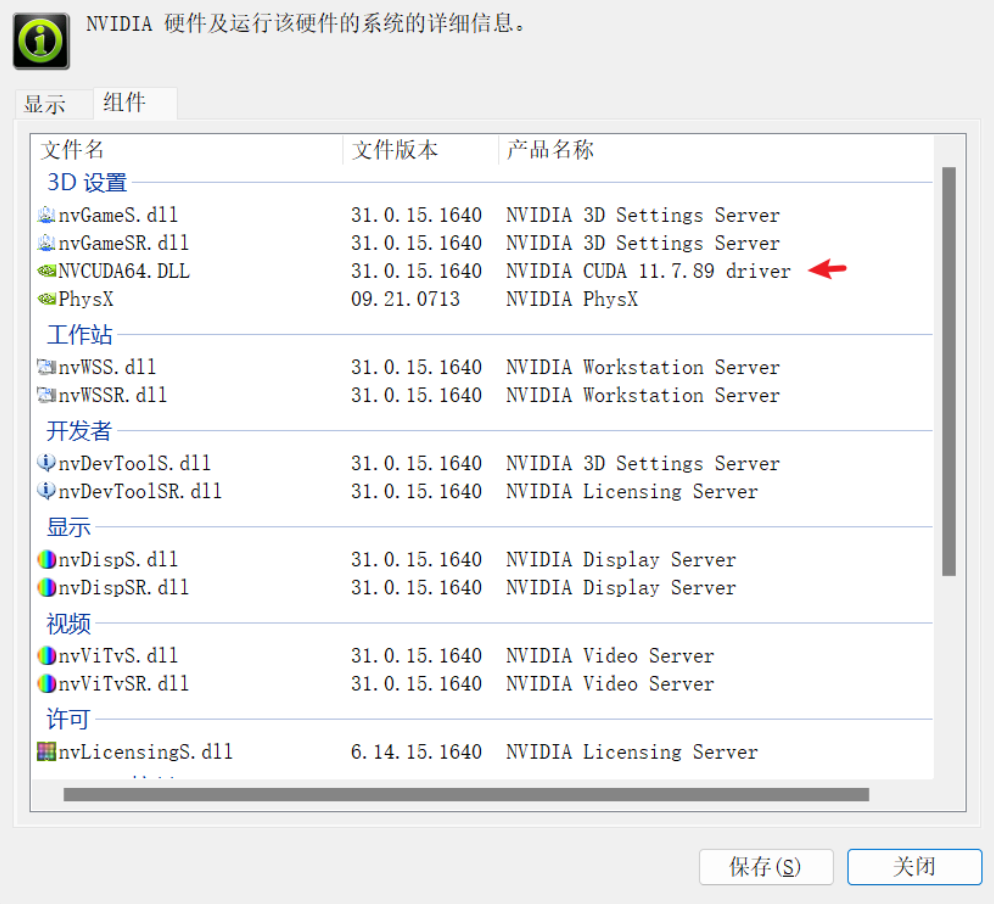
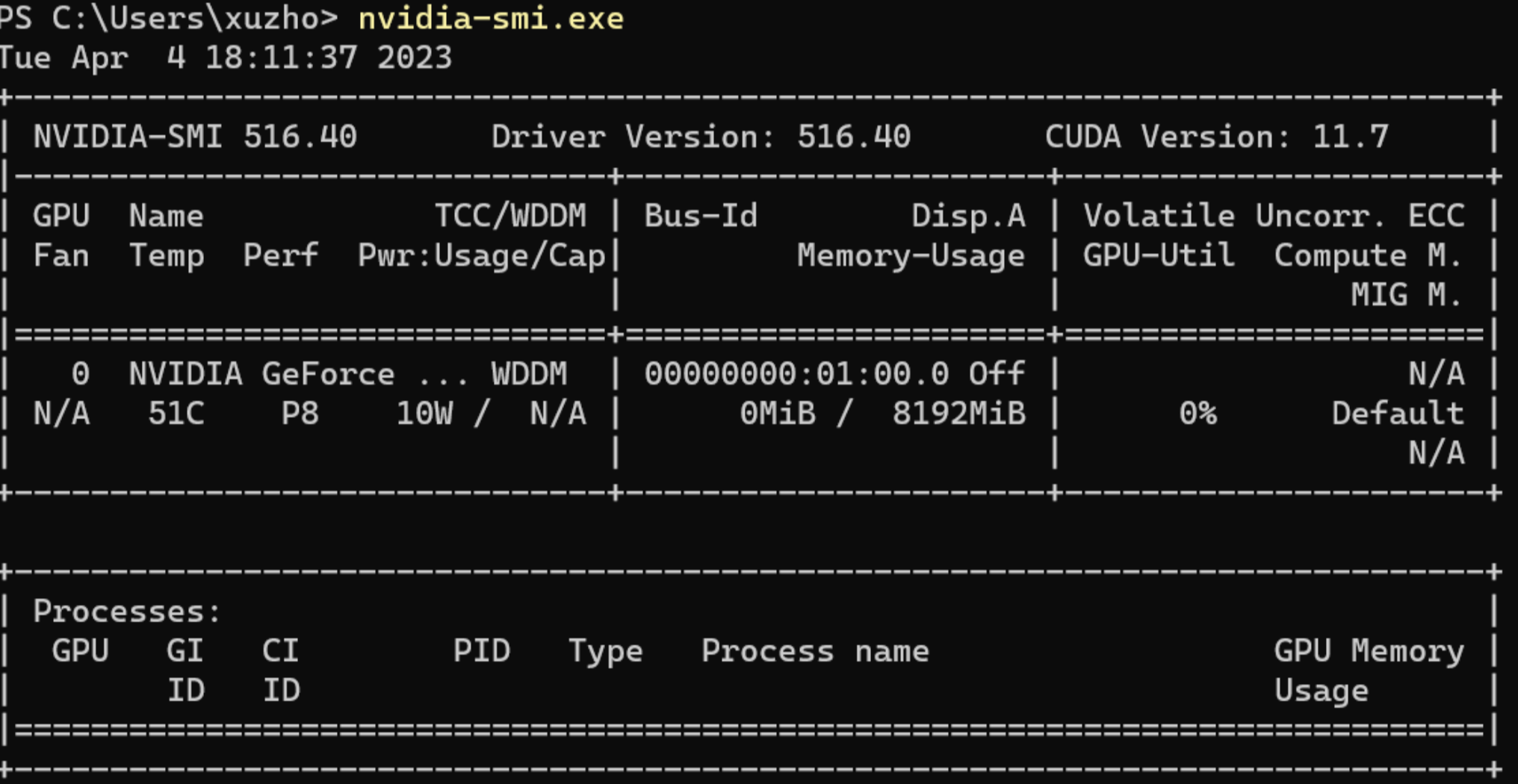
A: 我觉得这个跟装游戏类似,你虽然能装上游戏,但是不满足游戏的最低配置需求,照样跑不动。放在conda上,conda虽然能给你配置好cuda驱动,但是cuda驱动本身对系统的显卡驱动有要求。如果你的显卡驱动不满足,那么就算装好了,也会报错。RuntimeError: The NVIDIA driver on your system is too old
defforward(self, x): x = self.pool(torch.nn.functional.relu(self.conv1(x))) x = self.pool(torch.nn.functional.relu(self.conv2(x))) x = x.view(-1, 16 * 4 * 4) x = torch.nn.functional.relu(self.fc1(x)) x = torch.nn.functional.relu(self.fc2(x)) x = self.fc3(x) return x
net = Net()
# 这里的代码比较随意,就是用哪个平台运行哪个 # CPU device = torch.device("cpu") # CUDA device = torch.device("cuda:0") # MPS device = torch.device("mps")
/* F Format: use "F" format WHENEVER we use not more space than 'E' * and still satisfy 'R_print.digits' {but as if nsmall==0 !} * * E Format has the form [S]X[.XXX]E+XX[X] * * This is indicated by setting *e to non-zero (usually 1) * If the additional exponent digit is required *e is set to 2 */ ... *d = mxns - 1; *w = neg + (*d > 0) + *d + 4 + *e; /* width for E format */ if (wF <= *w + R_print.scipen) { /* Fixpoint if it needs less space */ *e = 0; if (nsmall > rgt) { rgt = nsmall; wF = mxsl + rgt + (rgt != 0); } *d = rgt; *w = wF; } /* else : "E" Exponential format -- all done above */ ...
# 如果翻译信息不同,则更新qualifiers字段 if new_translation_str != old_translation: feature.qualifiers["translation"] = [new_translation_str]
return record
if __name__ == '__main__': # 解析命令行参数 parser = argparse.ArgumentParser(description="Update CDS translations in a GenBank file.") parser.add_argument("input", help="Input GenBank file name") parser.add_argument("output", help="Output GenBank file name") parser.add_argument("-p", "--positions", help="New start and end positions for each gene, e.g. 'gene1:100-500 gene2:200-600'", default="") args = parser.parse_args()
# 读取GenBank文件和新位置信息 record = SeqIO.read(args.input, "genbank") new_positions = {gene_pos.split(":")[0]: tuple(map(int, gene_pos.split(":")[1].split("-"))) for gene_pos in args.positions.strip().split()}
# 如果翻译信息不同,则更新qualifiers字段 if new_translation_str != old_translation: feature.qualifiers["translation"] = [new_translation_str]
# 更新位置信息 feature.location = new_location
return record
if __name__ == '__main__': # 解析命令行参数 parser = argparse.ArgumentParser(description="Update CDS translations in a GenBank file.") parser.add_argument("input", help="Input GenBank file name") parser.add_argument("output", help="Output GenBank file name") parser.add_argument("-p", "--positions", help="New start and end positions for each gene, e.g. 'gene1:100-500 gene2:200-600'", default="") args = parser.parse_args()
# 读取GenBank文件和新位置信息 record = SeqIO.read(args.input, "genbank") new_positions = {gene_pos.split(":")[0]: tuple(map(int, gene_pos.split(":")[1].split("-"))) for gene_pos in args.positions.strip().split()}
defupdate_cds_translation(record, new_positions): """ 更新GenBank记录中所有CDS区域的翻译信息并返回修改后的记录。 :param record: GenBank记录对象 :param new_positions: 包含每个CDS区域新起始和终止位置的dict对象 :return: 修改后的GenBank记录对象 """ # 遍历每个特征 for feature in record.features: # 如果是CDS区域 if feature.type == 'CDS': # 获取特征的名称 gene = feature.qualifiers.get('gene', [''])[0] # 获取CDS区域的起始和终止位置 start, end = feature.location.start, feature.location.end # 如果有新位置信息,则使用新位置信息 if gene in new_positions: start, end = new_positions[gene] # 获取CDS区域的序列 cds_seq = record.seq[start:end] # 判断序列的方向 if feature.strand == -1: cds_seq = cds_seq.reverse_complement()
# 获取codon_start和transl_table信息 codon_start = int(feature.qualifiers.get('codon_start', [1])[0]) transl_table = int(feature.qualifiers.get('transl_table', [1])[0]) # 翻译CDS区域 table = Data.CodonTable.unambiguous_dna_by_id[transl_table] try: cds_protein = cds_seq[codon_start - 1:].translate(table=table, cds=True) except Data.CodonTable.TranslationError: cds_protein = '' print(f"Warning: Unable to translate CDS region {gene} in record {record.id}") # 更新CDS区域的翻译信息 feature.qualifiers['translation'] = [str(cds_protein)] return record
if __name__ == '__main__': # 解析命令行参数 parser = argparse.ArgumentParser(description="Update CDS translations in a GenBank file.") parser.add_argument("input", help="Input GenBank file name", type=argparse.FileType('r')) parser.add_argument("output", help="Output GenBank file name", type=argparse.FileType('w')) parser.add_argument("-p", "--positions", help="New start and end positions for each gene, e.g. 'gene1:100-500 gene2:200-600'", default="") args = parser.parse_args()
# 读取GenBank文件和新位置信息 records = (record for record in SeqIO.parse(args.input, "genbank")) new_positions = {gene: tuple(map(int, pos.split('-'))) for gene, _, pos inmap(str.partition, args.positions.split())} # 更新CDS翻译信息并写回GenBank文件 untranslated_cds_count = 0 for record in records: record = update_cds_translation(record, new_positions) # 统计无法翻译的CDS区域个数 for feature in record.features: if feature.type == 'CDS'andnot feature.qualifiers.get('translation', [''])[0]: untranslated_cds_count += 1 SeqIO.write(record, args.output, "genbank") # 提示用户有多少个CDS区域无法翻译为蛋白质序列 if untranslated_cds_count > 0: print(f"Warning: Failed to translate {untranslated_cds_count} CDS regions.")
我运行了这个代码,发现结果非常好。
1 2 3
$ python remake.py mitoscaf.fa.gbf new.gff Warning: Unable to translate CDS region COX2 in record XZG Warning: Failed to translate 1 CDS regions.
不同平台有着不同的Make工具用于编译,例如 GNU Make ,QT 的 qmake ,微软的 MS nmake,BSD Make(pmake),Makepp,等等。这些 Make 工具遵循着不同的规范和标准,所执行的 Makefile 格式也千差万别。这样就带来了一个严峻的问题:如果软件想跨平台,必须要保证能够在不同平台编译。而如果使用上面的 Make 工具,就得为每一种标准写一次 Makefile ,这将是一件让人抓狂的工作。
CMake就是针对上面问题所设计的工具:它首先允许开发者编写一种平台无关的 CMakeList.txt 文件来定制整个编译流程,然后再根据目标用户的平台进一步生成所需的本地化 Makefile 和工程文件,如 Unix 的 Makefile 或 Windows 的 Visual Studio 工程。从而做到“Write once, run everywhere”。显然,CMake 是一个比上述几种 make 更高级的编译配置工具。一些使用 CMake 作为项目架构系统的知名开源项目有 VTK、ITK、KDE、OpenCV、OSG 等.
1 2 3
wget https://cmake.org/files/v3.10/cmake-3.10.2.tar.gz tar xf cmake-3.10.2.tar.gz cd cmake-3.10.2
wget ftp://ftp.invisible-island.net/ncurses/ncurses.tar.gz && tar -zxvf ncurses.tar.gz ./configure --enable-shared --prefix=$HOME/usr make && make install
# libevent cd src wget https://github.com/libevent/libevent/releases/download/release-2.1.8-stable/libevent-2.1.8-stable.tar.gz tar -zxvf libevent-2.1.8-stable.tar.gz && cd libevent-2.1.8 ./configure prefix=$HOME/usr && make && make install
bzip2, xz, zlib: 文件压缩相关函数库,后续samtools编译时需要。
1 2 3 4 5 6
wget http://www.zlib.net/zlib-1.2.11.tar.gz tar -zxvf zlib-1.2.11.tar.gz && cd zlib-1.2.11 && ./configure --prefix=$HOME/usr && make && make install wget http://www.bzip.org/1.0.6/bzip2-1.0.6.tar.gz tar -zxvf bzip2-1.0.6.tar.gz && cd bzip2-1.0.6 && ./configure --prefix=$HOME/usr && make && make install wget https://tukaani.org/xz/xz-5.2.3.tar.gz tar -zxvf xz-5.2.3.tar.gz && cd xz-5.2.3 && ./configure --prefix=$HOME/usr && make && make install
wget http://ftp.gnu.org/gnu/readline/readline-7.0.tar.gz tar -zxvf readline-7.0.tar.gz && cd readline-7.0 ./configure --prefix=$HOME/usr && make && make install
PCRE: 提供和Perl5相同语法和语义正则表达式的函数库,后续安装R用到。
1 2 3
wget ftp://ftp.csx.cam.ac.uk/pub/software/programming/pcre/pcre-8.41.tar.gz tar -zxvf pcre-8.41.tar.gz && cd pcre-8.41 ./configure --enable-utf --enable-pcregrep-libz --enable-pcregrep-libbz2 --prefix=$HOME/usr
# 安装X11 wget -4 https://www.x.org/releases/X11R7.7/src/lib/libX11-1.5.0.tar.gz tar -zxvf libX11-1.5.0.tar.gz && cd libX11-1.5.0 ./configure --prefix=$HOME/usr && make && make install
wget -O zsh.tar.gz https://sourceforge.net/projects/zsh/files/latest/download tar -zxvf zsh.tar.gz && cd zsh export CPPFLAGS="-I$HOME/usr/include/" LDFLAGS="-L$HOME/usr/lib" ../configure --prefix=$HOME/usr --enable-shared make && make install
export CPPFLAGS="-I$HOME/usr/include -I$HOME/usr/include/ncurses" export LDFLAGS="-L$HOME/usr/lib -L$HOME/usr/lib64" mkdir -p src && cd src git clone https://github.com/tmux/tmux.git cd tmux sh autogen.sh ./configure --prefix=$HOME/usr make && make install