这篇是使用inferCNV分析单细胞转录组中拷贝数变异的后续,关于inferCNV在鉴定CNV上的一些思考。
用单细胞转录组分析CNV最早应该是2014年发表在science上的文章”Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma”
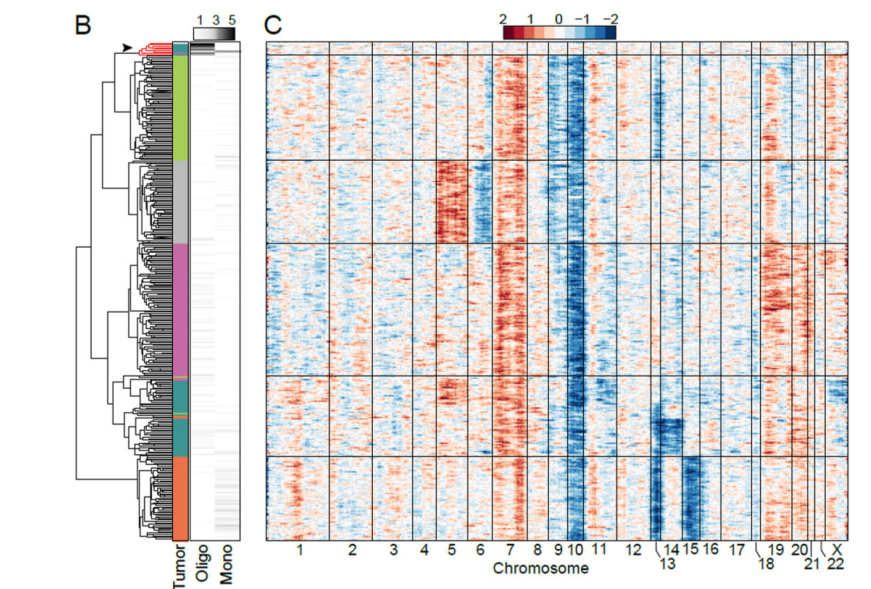
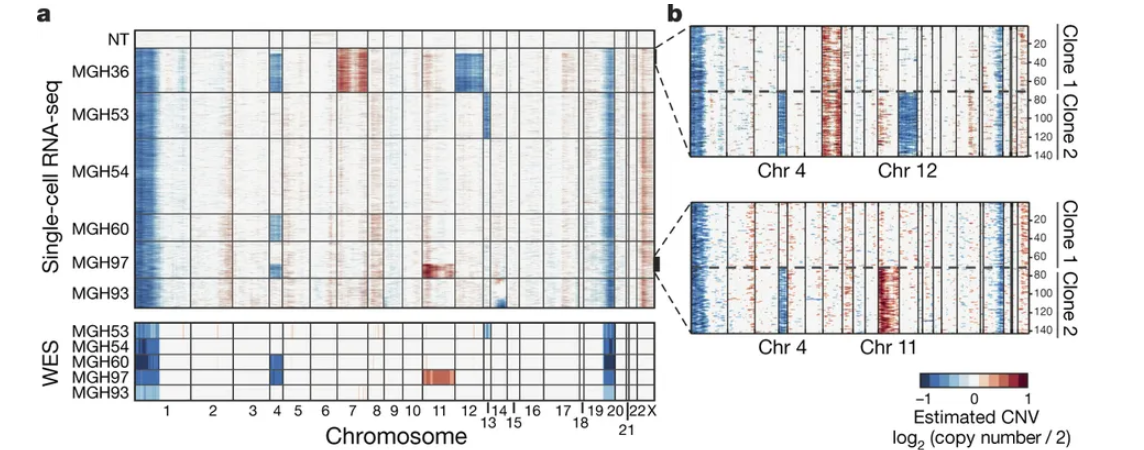
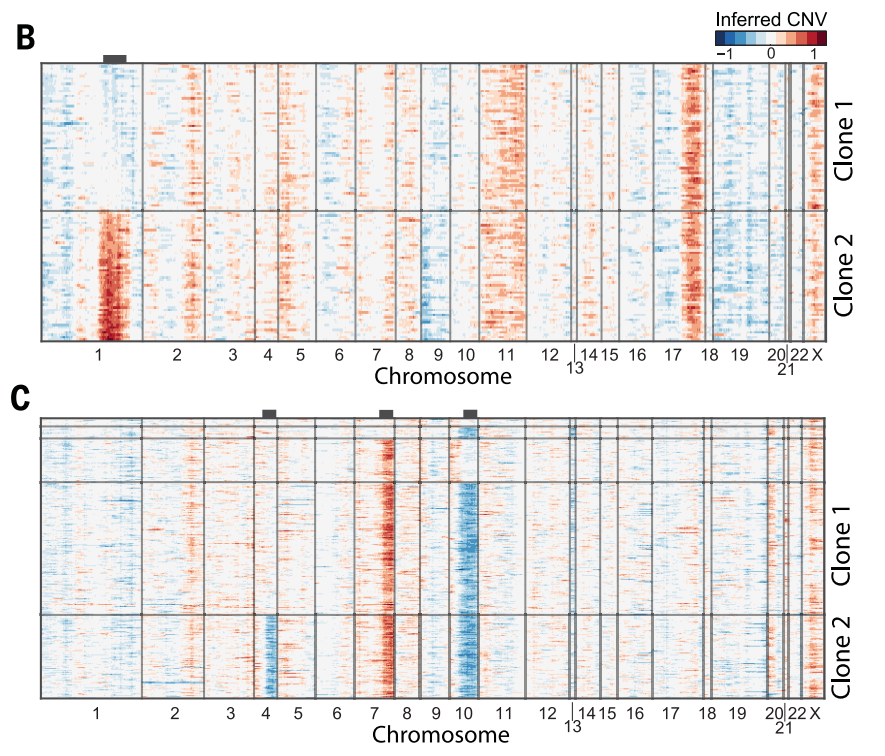
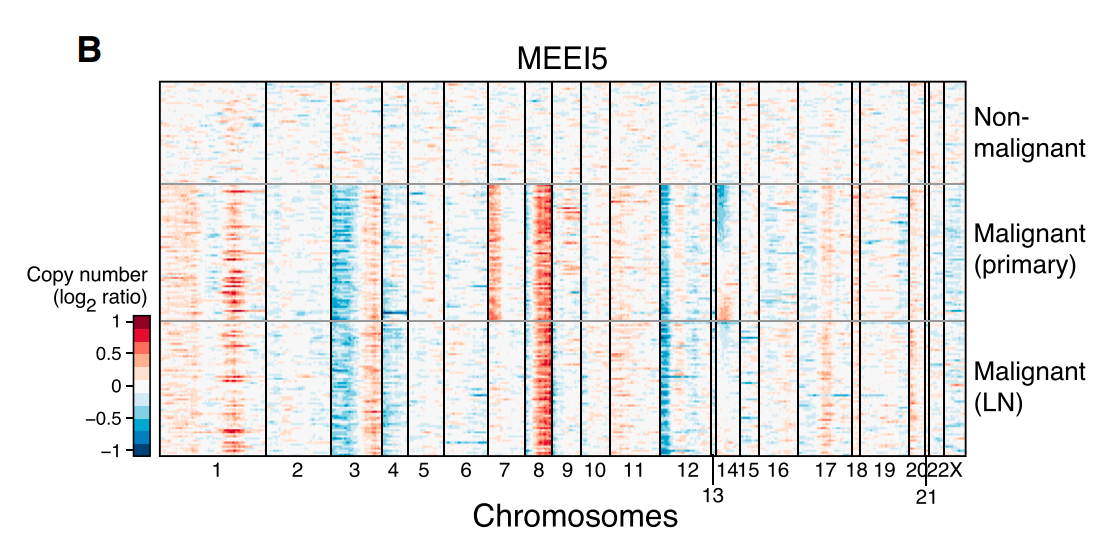
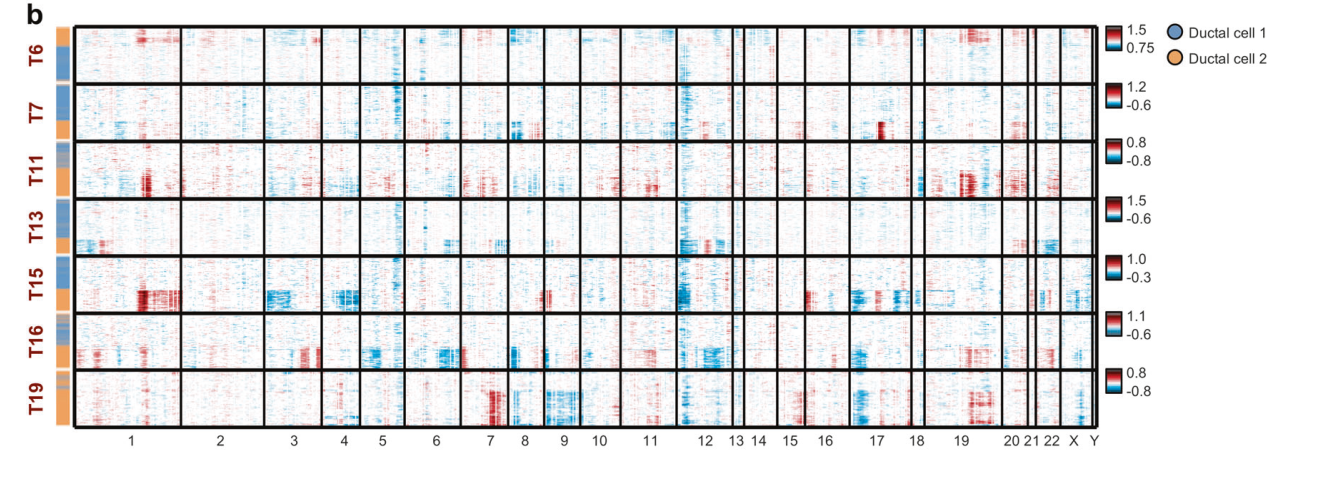
这篇是使用inferCNV分析单细胞转录组中拷贝数变异的后续,关于inferCNV在鉴定CNV上的一些思考。
用单细胞转录组分析CNV最早应该是2014年发表在science上的文章”Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma”
外周血单核细胞(Peripheral blood monoculear cell, PBMC), 包括淋巴细胞(T细胞,B细胞和自然杀伤(NK)细胞)和单核细胞。而红细胞和血小板没有细胞核,而粒细胞(granulocytes) 包括中性粒细胞、嗜碱性粒细胞和嗜酸性粒细胞,有多叶核,所以不包括在PBMC中。
人类的PBMC主要由淋巴细胞组成,之后是单核细胞以及一小部分的树突状细胞(dendritic cells, DC).
PBMC的提取方法: 利用Ficoll将全血分层,之后进行梯度离心, 血液就会分离成一层血浆,然后是一层PBMCs和一层多形核细胞(polymorphonuclear ),如中性粒细胞和嗜酸性粒细胞,和底层红细胞。通过溶解红细胞可以进一步分离多形核细胞。嗜碱性粒细胞有时出现在密度较高的组分中和PBMC组分中。
注: Ficoll: 一种亲水性多糖(hydrophillic polysaccharide )
PBMC起源于位于骨髓(bone marrow)的造血干细胞( hematopoietic stem cells, HSCs)。在造血干细胞的造血过程中,它们会产生髓系细胞(单核细胞、巨噬细胞、粒细胞、巨核细胞、树突状细胞、红细胞)和淋巴系细胞(T细胞、B细胞、NK细胞)。
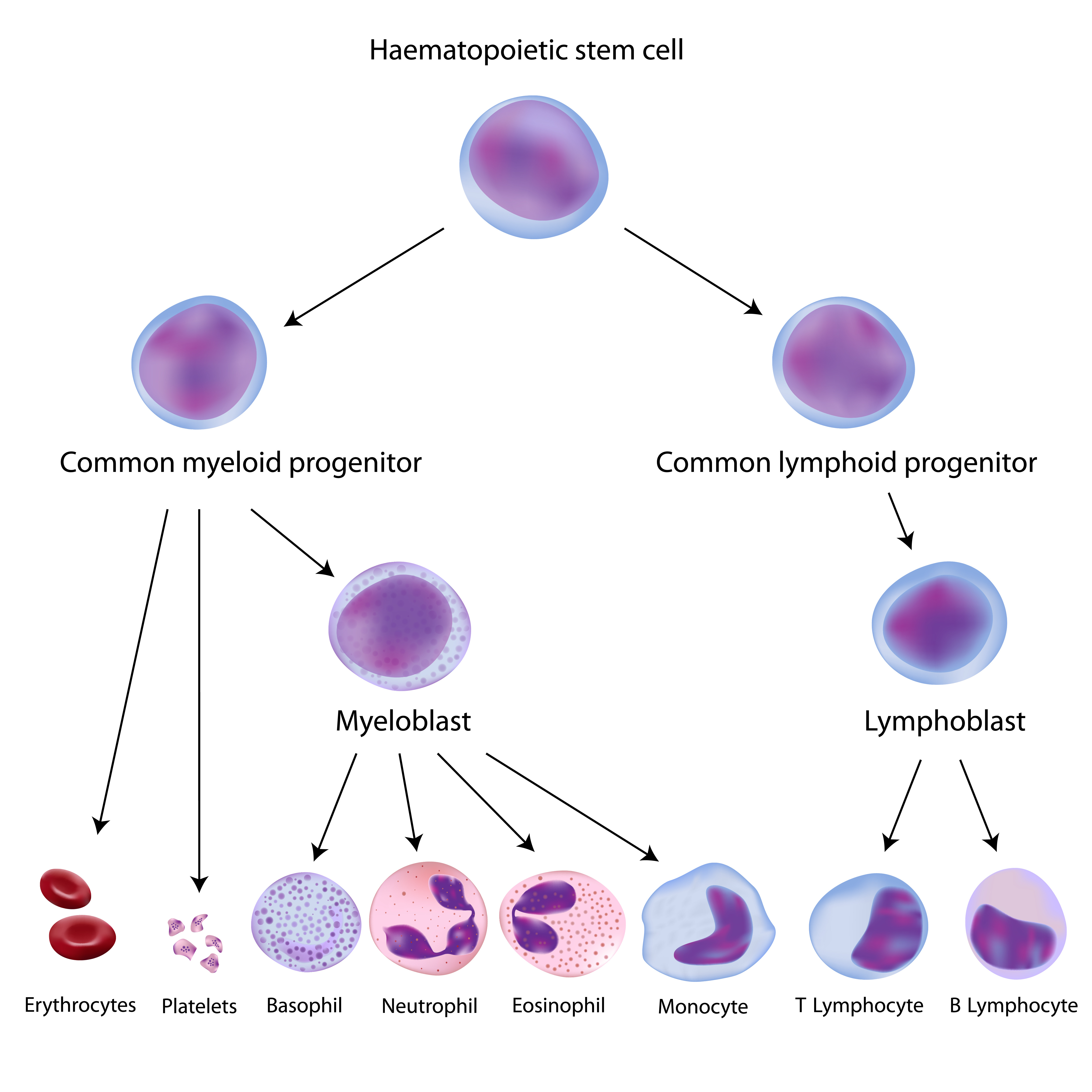
图解:
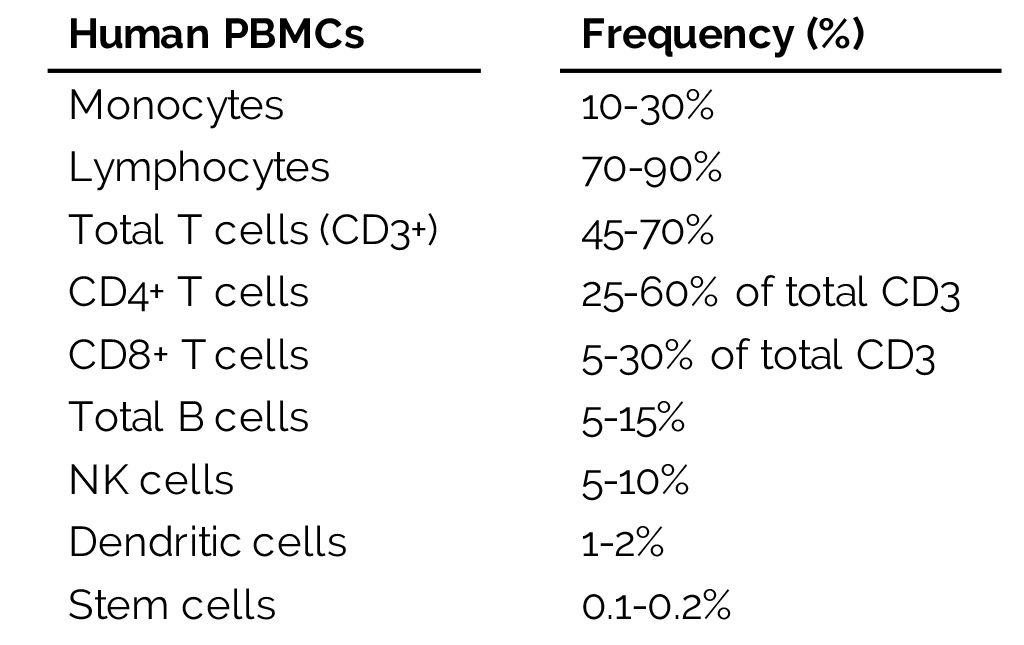
PBMC中大部分都是淋巴细胞,包括B细胞和T细胞,其中CD3+ T细胞又占了淋巴细胞中绝大部分(45-70%)。这些T细胞都处于初始(naive)状态,即已经成熟了但没有受到抗原刺激。 在正常人中,只有非常小的一部分初始T细胞会因抗原识别而被激活。同样的B细胞也都处于初始状态。
相对于淋巴细胞,单核细胞的比例在10-30%,收到刺激之后会发育成树突状细胞(DC)或巨噬细胞。
干细胞的比例非常低,只有0.1-0.2%,因此很难从全血样本中分离到。分子标记是CD34+。
https://en.wikipedia.org/wiki/Peripheral_blood_mononuclear_cell
https://www.stemexpress.com/blogs/peripheral-blood-mononuclear-cells/
我的生物信息学技能基本上都是自学的,在刚开始的阶段我也和大部分的人一样,会遇到很多问题,然后想着去一些群里提问希望得到解答,当然结果也是和大部分人的一样,问题不会得到答案。
后来,我通过看书和搜索慢慢的有了一些基础,通过项目实践对生信有了一点了解,加上自己也喜欢把自己的学习笔记公开出来,就逐渐有人开始向我提问。有些问题我会想去回答,而有些问题会被我直接忽视。如同当年我提出的问题一样,有些能够有人解答,而有些则石沉大海。我稍微的总结了一些,如果你希望让我或者别人回答你的问题,或许可以参考一下。
以我自己为例吧,我每天都会遇到很多问题,大部分的问题可以通过直接复制报错搜索的方式解决。有一些问题不是普遍性问题,我会通过提取报错中的关键信息进行检索。由于搜索的次数多了,我看到别人提问的时候,我差不多就能判断出这个问题需要经过几次搜索才能找到答案。对于能够通过简单搜索找到答案的问题,我就无视掉了。当然如果你在提问中指出你做了哪些搜索上的尝试,那么我会提供一些关键字,或者我会帮你去搜索下,如果发现是软件本身写的太差导致搜索不到的问题,那么我会建议你换个软件。
术业有专攻,没有经验就贸然回答,也是对提问人的不负责。比方说,我没有任何芯片数据处理的经验,也没有挖掘过GEO数据库或TCGA数据库。因此,当你问我如何合并不同平台的芯片数据时,我也只会说我不会,这不是推脱,是真的不会。
比方说一些工具类的报错,这些工具要么是我不会用到,要么是这个问题是某些平台所特有的,我很难重复出这个问题,那么我也不会花很多时间去重复这个问题。如果我以后遇到了这个问题,那么我会去解决,并给出解决方案。但是当下并不是我所在乎的,所以我就不会管它。
因为我一般会用手机看微信,如果我要回答你的问题,我就需要用手机打字。但问题手机的输入不如电脑的方便,而且容易出错,我会觉得很麻烦,就懒回答问题了。而我收邮件一般在电脑端,那么我用键盘打字就会很舒畅,当有些问题我也不太会的时候,也很容易搜索,我就不会感觉回答问题成为了一个负担,那么就会倾向于回答问题。
这类问题一个特征就是,它是一个选择题而不是一道简答题或者论述题。而且这类问题的描述一般会比较多,把问题的背景和自己的解答过程都写了出来,然后问你这样子做,合理吗?如果我觉得没问题,就只要回复一个“没问题”。如果我觉得有问题,那么我还会指出哪里不对,应该如何做。
以R语言为例吧,当你在某个问题出错时,如果能把代码和数据集都整理成一个压缩包,并且在出问题的地方,还加上提示,那么我就能很容易定位到问题,从而解决问题。
还有一类提问方式,就是知识付费形式的提问,这类问题大家都喜欢,你用自己辛辛苦苦学到的知识改善了自己的生活,这会让你感觉自己的努力没有白费。如果你以后在群里提问的时候附上红包,那么这个问题应该会有人看到吧。
最后,有问题的话可以发送邮件向我提问,至于我的邮箱地址,可以在我的简书上找到。
在MacOS或Linux上使用RStudio有一点不足,就是不能方便在RStudio上切换R版本。
虽然不方便,但还是可以切换。
首先,我们需要通过编译到方式安装不同版本的R,参考如何在服务器上安装最新的R
我习惯将软件安装在/opt/sysoft下
1 | sudo mkdir -p /opt/sysoft |
下载源代码,并安装
1 | wget https://mirrors.tuna.tsinghua.edu.cn/CRAN/src/base/R-3/R-3.5.3.tar.gz |
在安装完成之后,之后切换R语言,只需要用不同版本的R覆盖/usr/local/bin下面的R即可。
1 | ln -sf /opt/sysoft/R-3.5.3/bin/R /usr/local/bin |
之后打开RStudio就是更换版本后的R
inferCNV用与探索肿瘤单细胞RNA-seq数据,分析其中的体细胞大规模染色体拷贝数变化(copy number alterations, CNA), 例如整条染色体或大片段染色体的增加或丢失(gain or deletions)。工作原理是,以一组”正常”细胞作为参考,分析肿瘤基因组上各个位置的基因表达量强度变化. 通过热图的形式展示每条染色体上的基因相对表达量,相对于正常细胞,肿瘤基因组总会过表达或者低表达。
inferCNV提供了一些过滤参数,通过调整参数来降低噪音,更好的揭示支持CNA的信号。此外inferCNV还包括预测CNA区间的方法以及根据异质性模式定义细胞类群的方法。
尽管inferCNV是一个R包,但是在安装inferCNV之前还需要先下载安装JAGS ,好在它有Windows,MacOS和Linux版本,所以inferCNV在各个平台都能用。
Windows和MacOS的JAGS容易安装,而Linux的JAGS需要编译
1 | # 手动安装BLAS和LAPACK不推荐 |
安装R包
1 | install.packages("rjags") |
测试安装
1 | library(infercnv) |
如果没有报错,就说明安装成功。
需要准备3个输入数据
第一个是Genes x Cells的表达矩阵(matrix),行名是基因,列名是细胞编号。
| MGH54_P16_F12 | MGH54_P12_C10 | MGH54_P11_C11 | MGH54_P15_D06 | MGH54_P16_A03 | … | |
|---|---|---|---|---|---|---|
| A2M | 0 | 0 | 0 | 0 | 0 | … |
| A4GALT | 0 | 0 | 0 | 0 | 0 | … |
| AAAS | 0 | 37 | 30 | 21 | 0 | … |
| AACS | 0 | 0 | 0 | 0 | 2 | … |
| AADAT | 0 | 0 | 0 | 0 | 0 | … |
| … | … | … | … | … | … | … |
第二个是样本注释信息文件,命名为”cellAnnotations.txt”。一共两列,第一列是对应第一个文件的列名,第二列是细胞的分组
1 | MGH54_P2_C12 Microglia/Macrophage |
第三个是基因位置信息文件,命名为”geneOrderingFile.txt”。一共四列,第一列对应第一个文件的行名,其余三列则是基因的位置。注:基因名不能有重复
1 | WASH7P chr1 14363 29806 |
最复杂的工作就是准备输入文件,而一旦上述三个文件已经创建完成,那么分析只要两步以及根据结果对参数进行调整。
第一步,根据上述的三个文件创建inferCNV对象
1 | infercnv_obj = CreateInfercnvObject(raw_counts_matrix=matrix, # 可以直接提供矩阵对象 |
这一步的一个关键参数是ref_group_name, 用于设置参考组。假如你并不知道哪个组是正常,哪个组不正常,那么设置为ref_group_name=NULL, 那么inferCNV会以全局平均值作为基线,这适用于有足够细胞存在差异的情况。此外,这里的raw_count_matrix是排除了低质量细胞的count矩阵。
第二步,运行标准的inferCNV流程。
1 | # perform infercnv operations to reveal cnv signal |
关键参数是cutoff, 用于选择哪些基因会被用于分析(在所有细胞的平均表达量需要大于某个阈值)。这个需要根据具体的测序深度来算,官方教程建议10X设置为0.1,smart-seq设置为1。你可以先评估下不同阈值下的保留基因数,决定具体值。cluster_by_groups用于声明是否根据细胞注释文件的分组对肿瘤细胞进行分群。
最终会输出很多文件在out_dir的目录下,而实际有用的是下面几个
Infercnv::run的参数非常之多,总体上分为如下几类
你可以按照具体的需求修改不同步骤的参数,例如聚类默认cluster_by_groups=FALSE会根据k_obs_groups聚类成指定的组数,而层次聚类方法用于计算组间相似度的参数则是hclust_method.
此外,设置HMM=TRUE 的计算时间会长于HMM=FALSE,因此可以先设置HMM=FALSE快速查看结果。
在运行过程中它会显示每个步骤的信息,官方文档给出了示意图帮助理解。
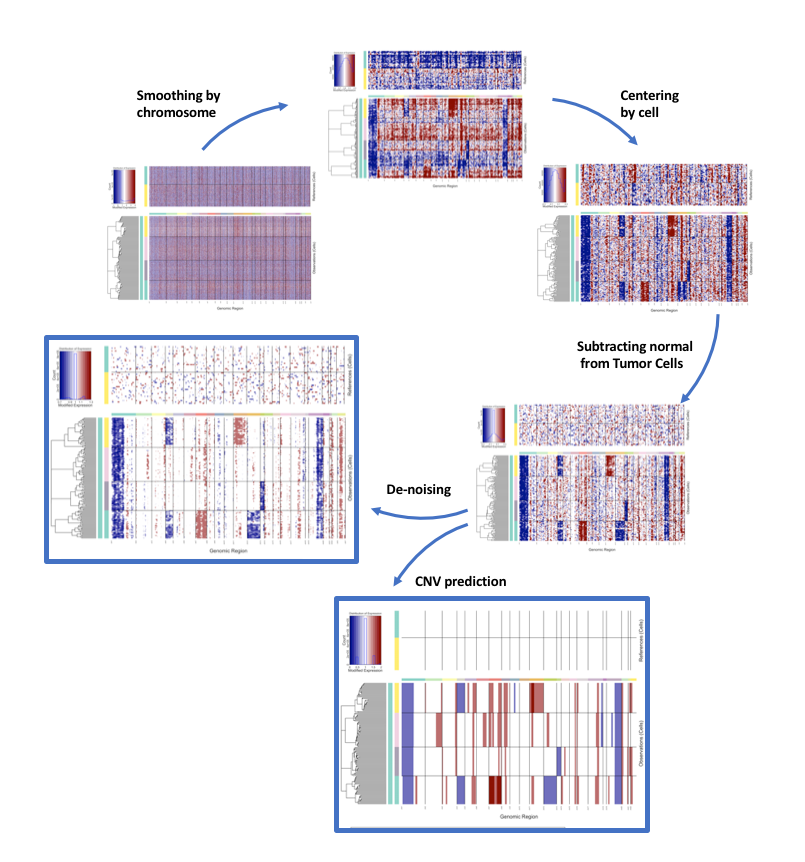
inferCNV会输出一个” map_metadata_from_infercnv .txt”文件用于记录每个细胞的元信息,所有信息都可以从该文件中进行提取。或者使用infercnv::add_to_seurat将信息直接增加到原来的seurat对象中。
关于inferCNV的算法原理在如下几篇文章中有说明
mclust(Model-based clustering) 能够基于高斯有限混合模型进行聚类,分类以及密度估计(density estimation)。对于具有各种协方差结构的高斯混合模型,它提供了根据EM算法的参数预测函数。它也提供了根据模型进行模拟的函数。还提供了一类函数,整合了基于模型的层次聚类,混合估计的EM算法,用于聚类、密度估计和判别分析中综合性策略的贝叶斯信息判别标准。最后还有一类函数能够对聚类,分类和密度估计结果中的拟合模型进行可视化展示。
简而言之,mclust在R语言上实现了基于高斯有限混合模型的聚类,分类和密度估计分析,并且还有专门的可视化函数展示分析结果。
和
mclust功能相似的其他R包: ‘Rmixmod’, ‘mixture’, ‘EMCluster’, ‘mixtools’, ‘bgmm’, ‘flexmix’
在已有的R语言的基础上,只需要运行如下代码即可
1 | # 安装 |
以一个例子来介绍一下如何使用mclust进行聚类分析。我们得要先加载一个来自于R包’gclus’的数据集’wine’,该数据集有178行,分别是不同区域的品种葡萄, 14列,其中后13列是化学分析的测量值。我们的目标是将其进行分类。
第一步: 加载数据集
1 | install.packages("gclus") |
第二步 : 使用Mclust做聚类分析. Mclust主要功能就是分析当前的提供的数据是由什么统计模型
1 | # 第一列和聚类无关 |
直接在交互行输入mod会得到如下信息
1 | 'Mclust' model object: (VVE,3) |
这里需要对结果稍作解释,第一行告诉我们’Mclust’以VVE模型将数据分为3类。第3行开始,它告诉我们’Mclust’的输出结果中包含了如下内容,我们可以通过$来提取。举个例子,我们提取Mclust的聚类结果和已知结果进行比较
1 | table(wine$Class, mod$classification) |
从结果中,我们发现仅有2例没有正确聚类,说明Mclust的效果很好。但是随之而来的问题是,Mclust如何挑选模型以及它为什么认为聚成3类比较合适呢?我们可以根据什么信息进行模型选择呢?
为了解答上面的问题,我们需要稍微了解点Mclust的原理。和其他基于模型的方法类似,Mclust假设观测数据是一个或多个混合高斯分布的抽样结果,Mclust就需要根据现有数据去推断最优可能的模型参数,以及是由 q几组分布抽样而成。mclust一共提供了14种模型(见下表),可以用?mclustModelNames可以查看mclust提供的所有模型。
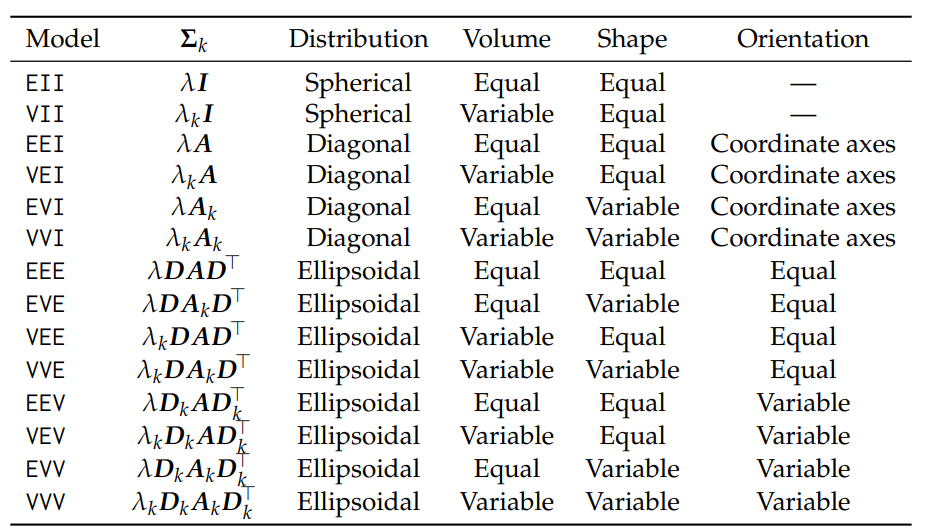
为了对模型有一个直观的理解,mclust提供了这些模型数据分为三组前提下在二维中的形状。
继续回到之前的问题,Mclust如何确定模型和确定分组数目。之前我们调用Mclust时,除了必须设置的输入参数,没有修改其他参数。其实Mclust可以设置的参数不少,和问题直接相关的是如下两个参数
1:9也就是,Mclust默认得到14种模型1到9组的分析结果,然后根据一定的标准选择最终的模型和分组数。
Mclust提供了两种方法用于评估不同模型在不同分组下的可能性
Mclust默认用的就是BIC,因此我们可以用plot.Mclust绘制其中BIC变化曲线
1 | plot.Mclust(mod, what = "BIC", |
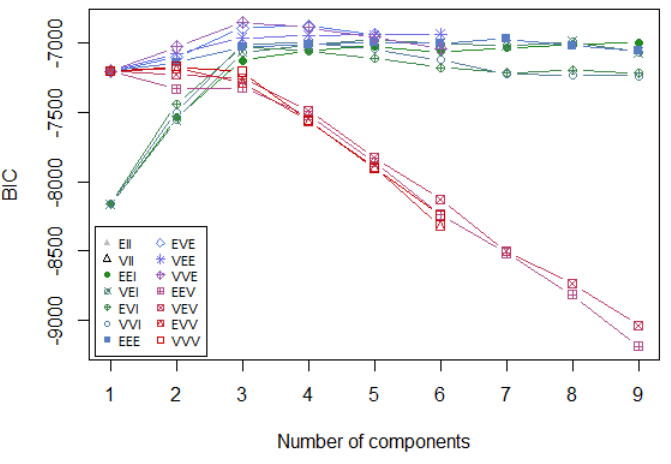
Mclucst会选择其中BIC最大的模型和分组作为最终的结果。
此外我们可以用MclustBIC和MclustICL分别进行计算
1 | par(mfrow=c(1,2)) |
从中选择最佳的模型分组和模型作为输入
1 | mod2 <- Mclust(X, G = 3, modelNames = "VVE", x=BIC) |
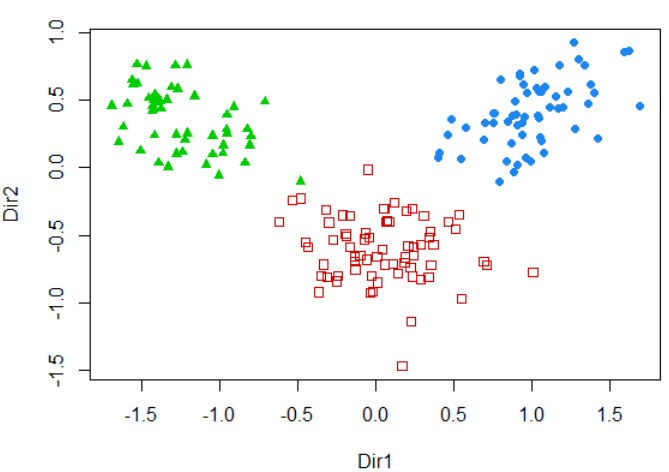
mclust为不同的输出都提供了对应的泛型函数用于可视化,你需要用plot就能得到结果。例如对之前的聚类结果在二维空间展示
1 | drmod <- MclustDR(mod, lambda = 1) |
mclust还有很多其他功能,例如密度估计,自举推断(Bootstrap inference),这些内容建议阅读”mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models “
想要更好的学习这个R包的使用,还需要去学习如下概念
在学习线性代数的时候,我们都会先从线性方程组入手。求解一个线性方程组是否存在解,如果存在解,那么有多少解,比方说求解下面这个方程组
$$
x_1 - 2x_2 = -1
$$
$$
-x1 - 2x_2 = 3
$$
先别急着掏出纸和笔进行运算。在线性代数中, 这类线性方程组更常见的表述方式为
$$
Ax = b
$$
其中,A是系数矩阵,x和b都是向量。在R语言中,这个方程组可以通过solve函数求解
1 | A <- matrix(c(1,-1,-2,-2), ncol = 2) |
如果方程组有解或者无数解,那么我们称方程组是相容的,反之则是不相容。
对于不相容的方程,我们无法求解出一个x使得等式成立,也就是下图的Ax和b不在一个平面上。因此只能找到一个x,让Ax尽可能b和接近,即求解一个x使得$||Ax-b||$最小。也就是$||Ax-b||^2$最小, 也就是$(Ax-b)\cdot(Ax-b)$最小, 这也就是术语最小二乘法( least squares method, 也称之为最小平方法 )的来源。
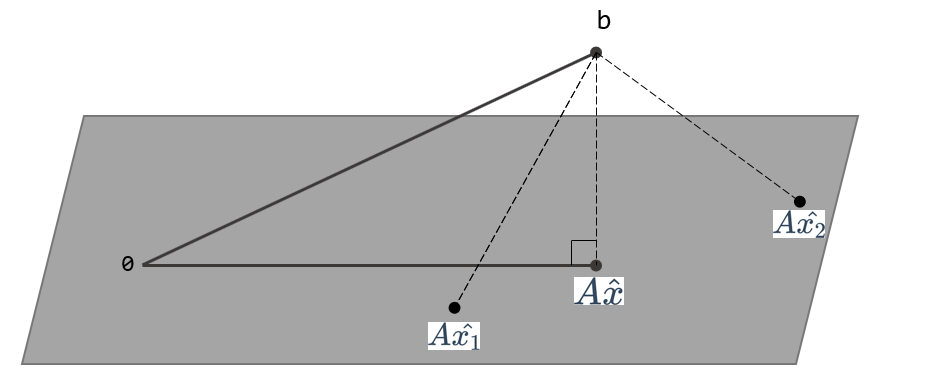
那么如何求解出x呢?跳过教科书的证明部分,这里直接提供定理,用于判定在什么条件下,方程$Ax=b$的最小二乘解是唯一的。

如果已知A的列是线性无关,那么对于方程$Ax=b$,取$A=QR$, 那么唯一的最小二乘解为$\hat x = R^{-1}Q^Tb$,
$A=QR$称之为QR分解,也就是将 $m \times n$ 矩阵A分解为两个矩阵相乘的形式。其中Q是 $m \times n$ 矩阵,其列形成$Col\ A$的一个标准正交基,R是一个 $n \times n$ 上三角可逆矩阵且在对角线元素为正数。
那么最小二乘有什么用呢?为了方便讨论实际的应用问题,我们用科学和工程数据中更常见的统计分析记号$X\beta=y$ 来替代$Ax=b$. 其中$X$为设计矩阵,$\beta$为参数向量,$y$为观测向量。
我们都知道身高和体重存在一定关系,但不是完美对应(数据集来自于R语言的women)
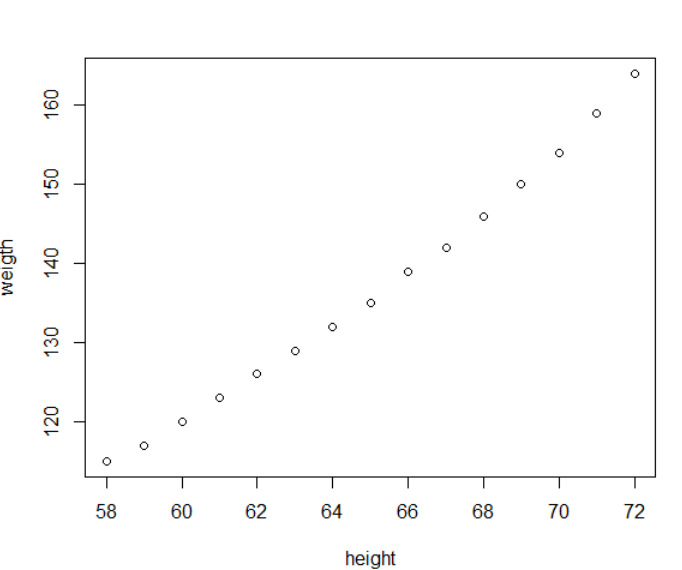
我们可以找到一条曲线去完美拟合已有的数据,但通常而言越复杂的模型就越容易出现过拟合的情况,不具有普适性,最好是找到一个简单模型,能对当前的数据做一个拟合,并且能预测未来。
两个变量最简单的关系就是线性方程$y=\beta_0 +\beta_1 x$。我们希望能够确定 $\beta_0$ 和 $\beta_1$使得预测值和观测值尽可能的接近,也就是让直线和数据尽可能的接近,最常用的度量方法就是余差平方之和最小。
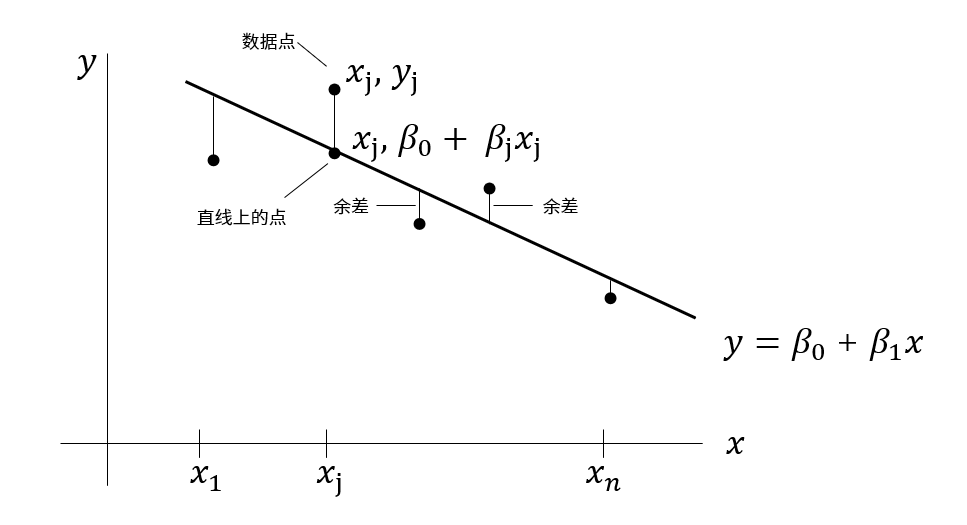
最小二乘直线$y=\beta_0 +\beta_1 x$是余差平方之和最小的,这条直线也被称之为y对x的回归直线。直线的系数 $ \beta_0$ 和 $\beta_1$称为线性回归系数。
如果数据点都落在直线上,那么参数 $ \beta_0$ 和 $\beta_1$满足如下方程,那么
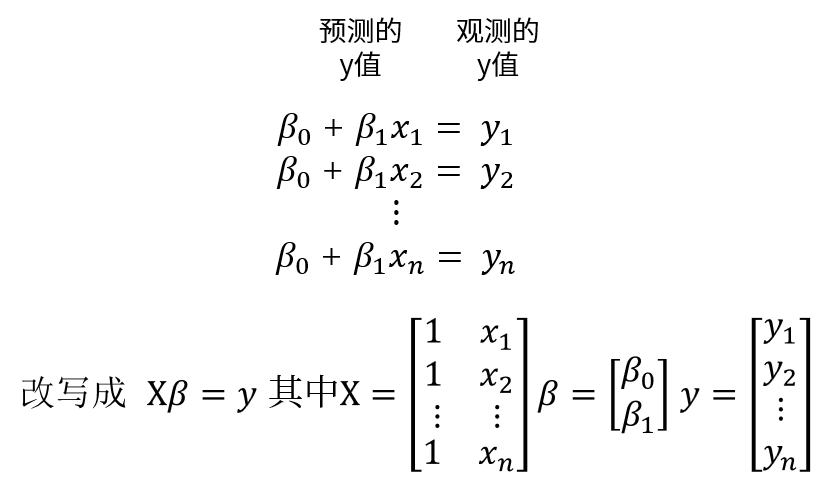
但是当数据点不在直线上时,这就意味着 $X\beta=y$ 没有解,那么问题其实是$Ax=b$的最小二乘问题,这两者仅仅记法不同。并且由于X的列线性无关,那么唯一的最小二乘解为$\hat \beta = R^{-1}Q^Ty$, 取$X=QR$,
接下来,我们就可以通过R语言的矩阵函数计算$\beta$
1 | # 加载数据 |
根据求解结果,系数分别是-87.52和3.45, 我们来绘图展示下
1 | plot(women$height, women$weight, |
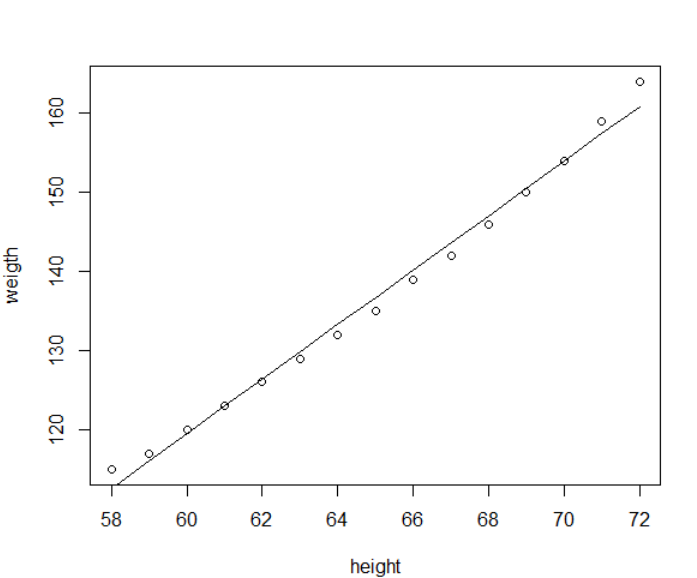
关于线性回归,R语言提供了lm函数,之前只是无脑调用,现在我把它的黑匣子打开了,它和我们之前做的事情类似,都有一步QR分解,只不过调用了不同的底层函数
1 | # lm |
lm的输出信息有很多,比如说残差,系数的显著性水平,这些可以用summary.lm查看。
之前看「R语言实战」的回归章节时,总是不理解,看完就记住了几个函数而已。现在在线性代数理论基础上对这个部分有所理解了。
如果对线性代数的最小二乘理论感兴趣,推荐阅读「线性代数及其应用」,戴维,原书第五版,第六章。
关于线性回归的R语言实现,推荐阅读「R语言实战」,第二版,第8章。
此外,维基百科的 最小二乘法页面也提供了比较好的解释。
在学习「数据挖掘导论」的数据预处理时,里面谈到了变量变换,我联想到了在基因表达量分析时的常见操作,例如FPKM,TPM,CPM,log对数变换。 比如说在文章里面会见到如下的描述
The size factor of each cell was computed using a pooling strategy implemented in the R function
computeSumFactors. Normalized counts were then computed by dividing the counts for each cell by the size factor for that cell. A log2 transformation was applied to normalized counts.
变量转换有什么好处,需要注意些什么呢?「数据挖掘导论」讨论了两种重要的变量变换类型: 简单函数变换和规范化。
简单变换是使用一个简单数学函数分别作用于每一个值,例如log转换,求绝对值,求倒数等。统计学中,变量变换(例如log转换)常用于将不具有高斯(正态)分布的数据变换成具有高斯(正态)分布的数据。在数据挖掘领域可以用来进行数据压缩。这类变换需要我们了解数据在变化前后的后果,例如负数取倒数之后的大小关系会逆转。
标准化(standardization)或规范化(normalization)的目标是使整个值的集合具有特定的属性。使用这两个术语需要特别注意使用这两个词的上下文。书中提到”在数据挖掘界,这两个术语常常可互换,然而,在统计学中,术语规范化可能与使得变量正态化相混淆”。虽然从中文的角度来看,规范化和正态化明显不一样,但是从英文的角度看,正态分布翻译自normal distribution, 很容易从normal联想到normalization。当然也有将normalization翻译成归一化,即将原来数据值通过$(x - min(X) / (max(X) - min(X))$转换到0,1之间。统计学的变量标准化指的就是对原来的数据基于均值和标准差计算z-score(公式如下), 不过考虑到均值和标准差受到离群点波动很大,可以用中位数替代均值,用绝对标准差替代标准差。
$$
x’ = \frac{x-\bar{x}}{\sigma}
$$
R语言中做标准化常用到一个函数scale,它的功能是对矩阵的列进行中心化和(或)缩放
1 | scale(x, center = TRUE, scale = TRUE) |
参数就3个,基本上我用的时候就提供第一个输入,举个例子
1 | > x <- c(1,2,3,4,5) |
之前我都是这样子用的,也没有思考过这到底它到底做了些什么。最近为了彻底搞懂它的两个参数,我翻了它的源代码。
代码总共38行,第一部分是关于center参数,也就是中心化。它先判断center是否是逻辑值还是数值向量。如果是逻辑值,则当center=TRUE时它会计算每一列的均值,然后调用sweep按列分别减去均值。如果判定center是一个数值向量时,就是为sweep手动指定每一列的中心值。
1 | if (is.logical(center)) { |
第二部分是关于scale,决定数据如何缩放。如果是逻辑值且为真时,默认用下面的函数计算每一列的缩放系数,然后用sweep按列除以每列系数
$$
\sqrt{\frac{\sum_{i=1}^{n}x{‘}_i^2}{n-1}}
$$
这个计算的结果就是样本标准差,因为$x’_i=x_i-\bar x$
如果是数值向量,则用sweep以给定数值按列相除。
1 | if (is.logical(scale)) { |
最后一部分是设置输出结果的属性。
1 | if(is.numeric(center)) attr(x, "scaled:center") <- center |
那么默认参数下,我们就是对矩阵按列进行z-score的标准化。检验标准很简单,计算标准化的数据的均值和标准差,因为z-score的结果就是通过线性变换让数据的均值为0,标准差为1(不会改变原来的数据分布)。
1 | > mean(y) |
这解决了我多年的疑惑,为啥我在R语言中就是找不到zscore这个函数,因为scale默认情况下就是实现z-score的变换。当然R语言取名scale也是有它的道理,比方说数据中存在离群值,我们应该选择不容易受离群值影响的中位数替代均值,选择绝对平均偏差, 中位数绝对偏差或四分位数极差来替换方差。
1 | # 默认参数 |
此外,它还能实现其他形式的数据转换,例如归一化,即将数据的范围缩放到0到1之间
1 | x <- matrix(c(1,2,3,4,5,6), ncol=2) |
最后总结一下:
scale默认就是计算原始数据的z-score, 通过调整center和scale可以实现多种形式的数据转换。
LACHESIS是一个比较早利用HiC数据辅助基因组组装的工具,文章发表在Nature Biotechnology上.但是这款软件在2年前就不在维护了,GitHub主页上也推荐使用https://github.com/theaidenlab里的工具进行组装。
不过目前依旧有很多公司在用这个软件做基因组组装,所以我也想试试看。
LACHESIS需要安装在Linux系统系统,最低内存不低于16GB,要求最小堆栈大小为10MB,可以用ulimit -s查看,通过ulimit -s 10240。同时依赖于以下软件
以没有root权限为例,安装SAMtools和Boost这两个依赖库
1 | mkdir ~/src |
然后是下载并解压缩
1 | cd ~/opt/biosoft/ |
由于之前安装的samtools并不是安装在/usr目录下,因此要修改src/include/gtools/目录下的SAMStepper.h和SAMStepper.cc中的#include <bam/sam.h>, 指向实际地址。例如我的是#include "/home/xzg/src/samtools-0.1.19/sam.h"
1 | export LACHESIS_BOOST_DIR=$HOME/src/boost_1_57_0 |
如果服务器管理员帮你安装了一个高版本的boost,你需要让他进行卸载,否则默认会线用系统的boost,然后就会出错。关于更多的报错,参考HiC软件安装篇之Lachesis
如果你Perl版本不是 5.14.2 ,那么我们需要打开bin下面的perl脚本,删除如下信息
1 | /////////////////////////////////////////////////////////////////////////////// |
而且还得将第一行替换成#!/usr/bin/perl -w
之后需要将LACHESIS加入环境变量
1 | export PATH=$PATH:~/biosoft/LACHESIS/bin |
为了保证软件在后续不会出错,我们还要测试下
1 | cd bin |
当你看到最后结果是: Done!就表明运行成功了,最终结果输出在当前目录下outs中
实际的软件使用要求你需要提供至少两类输入文件
参考使用ALLHiC基于HiC数据辅助基因组组装的第一步,第二步和第三步,使用bwa-aln + bwa-sampe将fastq回帖到参考基因组,然后对数据进行过滤。那么在后续的流程都假设你有如下两个文件
然后我们需要拷贝一个配置运行文件
1 | cp ~/opt/biosoft/LACHESIS/bin/INIs/test_case.ini sample.ini |
接着去修改参数,由于每个参数都会有说明,这里就展示我编辑过的几个参数
1 | SPECIES = test # 写实际的物种名即可 |
上面这些参数的修改属于基本操作,而下面的参数可能需要反复修改
1 | # contig中最小的酶切位点数, |
最终运行即可
1 | ulimit -s 10240 |
我们来构建最终的组装fasta
1 | CreateScaffoldedFasta.pl draft.asm.fasta out/test_case |
最终结果输出在out/test_case/Lachesis_assembly.fasta
使用体验:
目前的想法: 这个软件我不会再用第二次了。
ALLHiC除了能够解决复杂基因组(高杂合,多倍体)的基因组组装问题,还能够搞定简单的基因组组装,毕竟本质上都是利用HiC的信号。
ALLHiC大概有两个优势:
参考使用ALLHiC基于HiC数据辅助基因组组装完成软件安装和前三步的数据预处理,将Fastq文件转成BAM文件。
下面假定你的初步组装结果是draft.asm.fasta, Fastq预处理后的文件是sample.clean.bam
之后,我们将contig根据相互之间的信号强弱关系进行分组,至于分成多少组要根据物种具体的染色体数目而定。
1 | ALLHiC_partition -b sample.clean.bam -r draft.asm.fasta -e AAGCTT -k 16 |
要根据自己的实际建库方法选择酶切位点的识别序列,注, DpnI, DpnII, MboI, Sau3AI 这些酶都是识别相同的序列,仅仅是对甲基化敏感度不同。
接着,提取CLM和每个contig的酶切数
1 | allhic extract sample.clean.bam draft.asm.fasta --RE AAGCTT |
然后是对每个组的contig位置和方向进行优化
1 | # 如下命令可以通过循环同时运行 |
最终获取染色体级别的组装
1 | ALLHiC_build draft.asm.fasta |
绘制热图对组装进行评估
1 | # 获取染色体长度 |
其中getFaLen.pl可以从https://github.com/tangerzhang/my_script获取