# 如果翻译信息不同,则更新qualifiers字段 if new_translation_str != old_translation: feature.qualifiers["translation"] = [new_translation_str]
return record
if __name__ == '__main__': # 解析命令行参数 parser = argparse.ArgumentParser(description="Update CDS translations in a GenBank file.") parser.add_argument("input", help="Input GenBank file name") parser.add_argument("output", help="Output GenBank file name") parser.add_argument("-p", "--positions", help="New start and end positions for each gene, e.g. 'gene1:100-500 gene2:200-600'", default="") args = parser.parse_args()
# 读取GenBank文件和新位置信息 record = SeqIO.read(args.input, "genbank") new_positions = {gene_pos.split(":")[0]: tuple(map(int, gene_pos.split(":")[1].split("-"))) for gene_pos in args.positions.strip().split()}
# 如果翻译信息不同,则更新qualifiers字段 if new_translation_str != old_translation: feature.qualifiers["translation"] = [new_translation_str]
# 更新位置信息 feature.location = new_location
return record
if __name__ == '__main__': # 解析命令行参数 parser = argparse.ArgumentParser(description="Update CDS translations in a GenBank file.") parser.add_argument("input", help="Input GenBank file name") parser.add_argument("output", help="Output GenBank file name") parser.add_argument("-p", "--positions", help="New start and end positions for each gene, e.g. 'gene1:100-500 gene2:200-600'", default="") args = parser.parse_args()
# 读取GenBank文件和新位置信息 record = SeqIO.read(args.input, "genbank") new_positions = {gene_pos.split(":")[0]: tuple(map(int, gene_pos.split(":")[1].split("-"))) for gene_pos in args.positions.strip().split()}
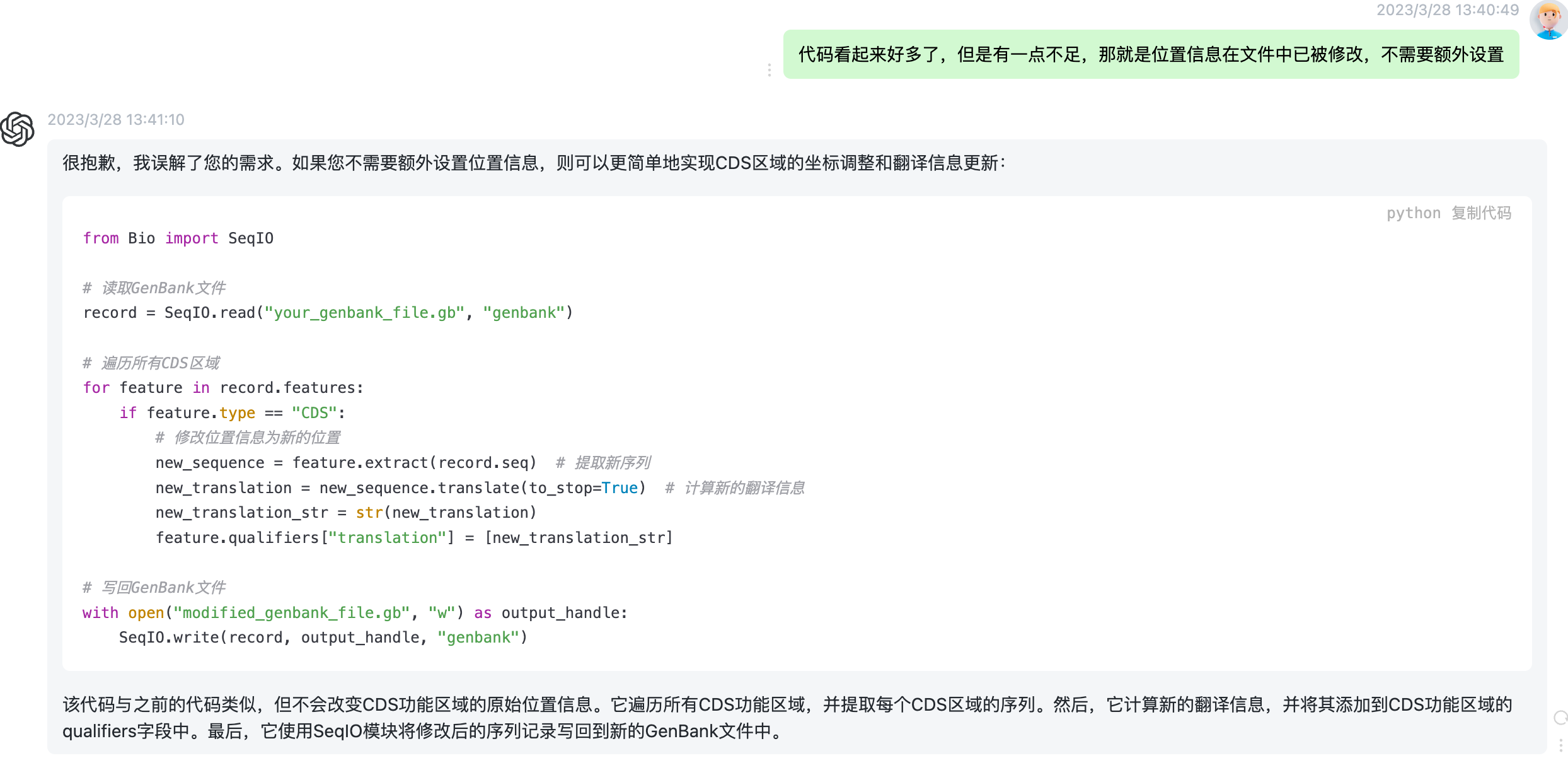
defupdate_cds_translation(record, new_positions): """ 更新GenBank记录中所有CDS区域的翻译信息并返回修改后的记录。 :param record: GenBank记录对象 :param new_positions: 包含每个CDS区域新起始和终止位置的dict对象 :return: 修改后的GenBank记录对象 """ # 遍历每个特征 for feature in record.features: # 如果是CDS区域 if feature.type == 'CDS': # 获取特征的名称 gene = feature.qualifiers.get('gene', [''])[0] # 获取CDS区域的起始和终止位置 start, end = feature.location.start, feature.location.end # 如果有新位置信息,则使用新位置信息 if gene in new_positions: start, end = new_positions[gene] # 获取CDS区域的序列 cds_seq = record.seq[start:end] # 判断序列的方向 if feature.strand == -1: cds_seq = cds_seq.reverse_complement()
# 获取codon_start和transl_table信息 codon_start = int(feature.qualifiers.get('codon_start', [1])[0]) transl_table = int(feature.qualifiers.get('transl_table', [1])[0]) # 翻译CDS区域 table = Data.CodonTable.unambiguous_dna_by_id[transl_table] try: cds_protein = cds_seq[codon_start - 1:].translate(table=table, cds=True) except Data.CodonTable.TranslationError: cds_protein = '' print(f"Warning: Unable to translate CDS region {gene} in record {record.id}") # 更新CDS区域的翻译信息 feature.qualifiers['translation'] = [str(cds_protein)] return record
if __name__ == '__main__': # 解析命令行参数 parser = argparse.ArgumentParser(description="Update CDS translations in a GenBank file.") parser.add_argument("input", help="Input GenBank file name", type=argparse.FileType('r')) parser.add_argument("output", help="Output GenBank file name", type=argparse.FileType('w')) parser.add_argument("-p", "--positions", help="New start and end positions for each gene, e.g. 'gene1:100-500 gene2:200-600'", default="") args = parser.parse_args()
# 读取GenBank文件和新位置信息 records = (record for record in SeqIO.parse(args.input, "genbank")) new_positions = {gene: tuple(map(int, pos.split('-'))) for gene, _, pos inmap(str.partition, args.positions.split())} # 更新CDS翻译信息并写回GenBank文件 untranslated_cds_count = 0 for record in records: record = update_cds_translation(record, new_positions) # 统计无法翻译的CDS区域个数 for feature in record.features: if feature.type == 'CDS'andnot feature.qualifiers.get('translation', [''])[0]: untranslated_cds_count += 1 SeqIO.write(record, args.output, "genbank") # 提示用户有多少个CDS区域无法翻译为蛋白质序列 if untranslated_cds_count > 0: print(f"Warning: Failed to translate {untranslated_cds_count} CDS regions.")
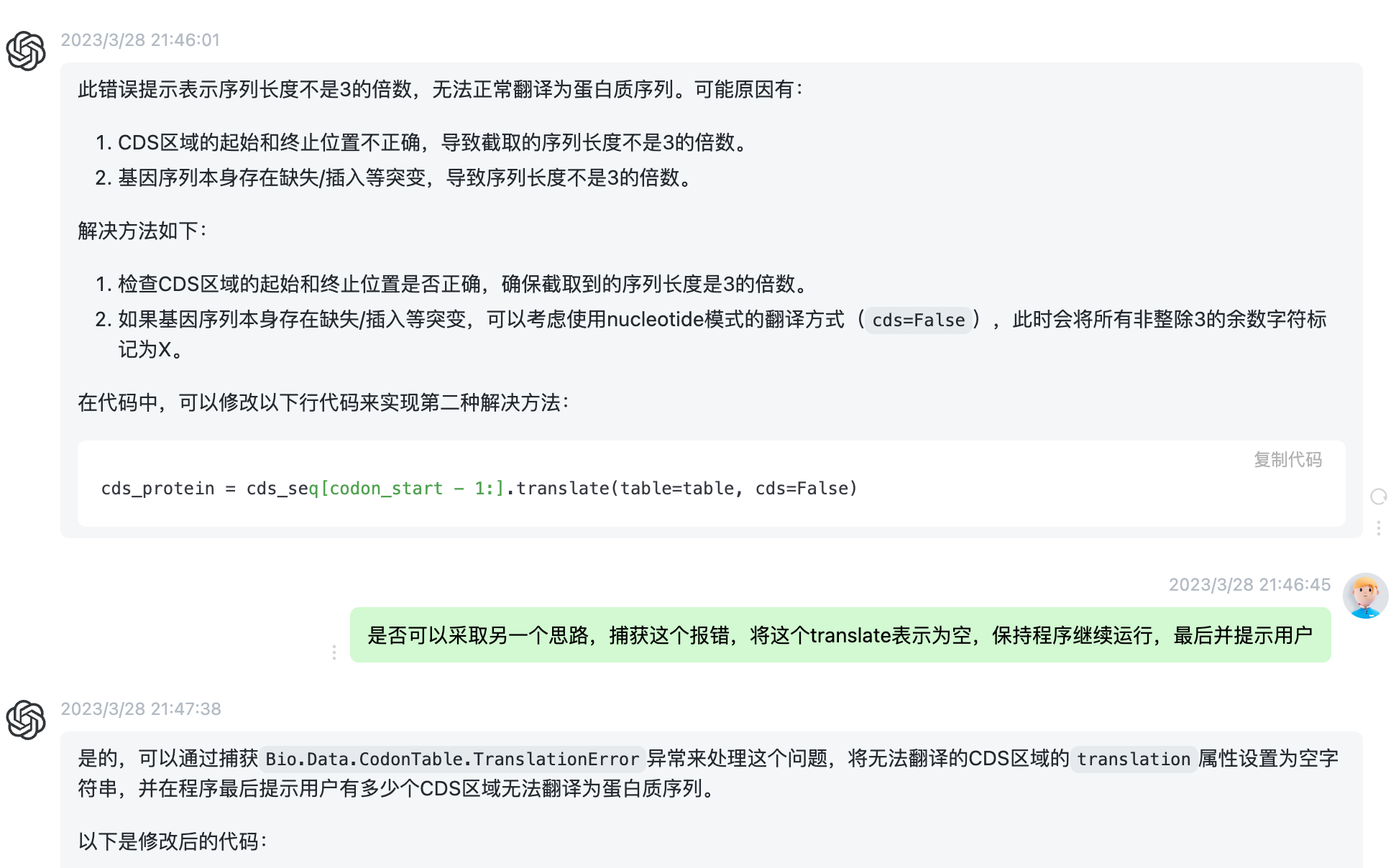
我运行了这个代码,发现结果非常好。
1 2 3
$ python remake.py mitoscaf.fa.gbf new.gff Warning: Unable to translate CDS region COX2 in record XZG Warning: Failed to translate 1 CDS regions.