blockinfo模块输出文件以csv格式进行存放,共23列,可以用EXCEL直接打开。
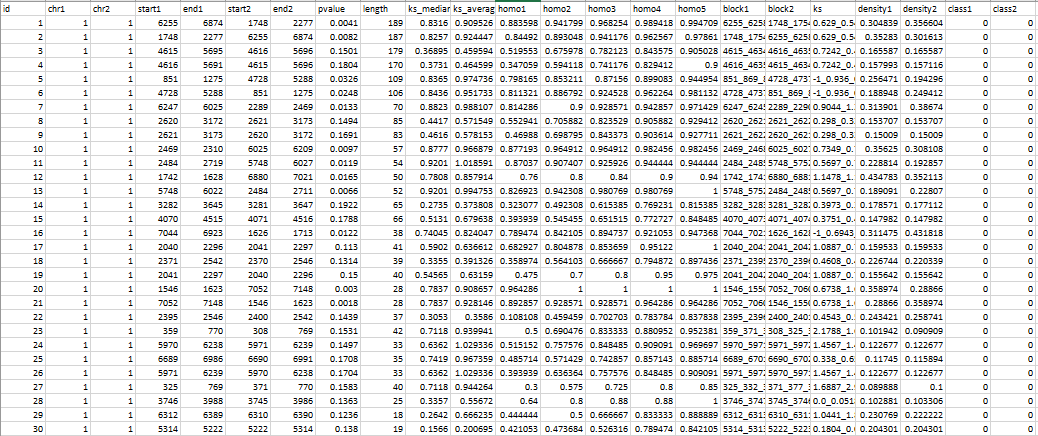
其中16列非常容易裂解,描述如下
id即共线性的结果的唯一标识chr1,start1,end1即参考基因组(点图的左边)的共线性范围(对应GFF1的位置)chr2,start2,end2即参考基因组(点图的上边)的共线性范围(对应GFF2的位置)pvalue即共线性结果评估,常常认为小于0.01的更合理些length即共线性片段中基因对数目ks_median即共线性片段上所有基因对ks的中位数(主要用来评判ks分布的)ks_average即共线性片段上所有基因对ks的平均值block1,block2分别为共线性片段上基因order的位置。ks共线性片段上所有基因对的ks值density1,density2共线性片段的基因分布密集程度。值越小表示稀疏。
最后两列,class1和 class2会在 alignment 模块中用到,对应的是两个block分组,默认值是0表示两个block是同一组。这两列后期需要自己根据覆盖率,染色体核型等多个方面进行确定。举个例子,我们可以根据 homo1 的取值范围对class1进行赋值,例如-1-0.5 是 1,-0.5 ~ 0.5 是2,0.51是3,最后在alignment中会就会用三种颜色来展示,例如下图的1,2,3分别对应red,blue,green.

中间的homo1,homo2,homo3,homo4,homo5并非那么直观,先说结论:
这里的homoN(N=1,2,3,4,5) 表示一个基因有N个最佳匹配时的取值
N由mutiple参数确定,对应点阵图(dotplot)中的红点
multiple的取值一般取1即可,表示最近一次的WGD可能是一次二倍化事件,因此每个基因只会有一个最佳匹配。如果设置为2,可能是一次3倍化,每个基因由两个最佳匹配。当然实际情况可能会更加复杂,比如说异源四倍体,或者异源六倍体,或者没有多倍化只是小规模的基因复制(small-scale gene duplication) 等情况,也会影响multiple的设置。
homoN会在后面过滤共线性区块时用到,一般最近的WGD事件所产生的共线性区块会比较接近1,而古老的WGD产生的共线性区块则接近-1.
接着,我们将根据源代码 blast_homo和blast_position 来说明结算过程。
首先需要用到blast_homo函数,用来输出每个基因对在不同最佳匹配情况下的取值(-1,0,1)。
1 | def blast_homo(self, blast, gff1, gff2, repeat_number): |
for循环前的代码作用是提取每个基因BLAST后的前N个最佳结果。循环的作用基因对进行赋值,主要规则是基因对如果在点图中为红色,赋值为1,蓝色赋值为0,灰色赋值为-1。
homo1 对应 redindex = 0:1, bluenum = 1:6, grayindex = 6:repeat_number
homo2 对应redindex = 0:2, bluenum = 2:7, grayindex = 7:repeat_number
…
homo5对应redindex=0:5, bluenum=5:10, grayindex = 10:repeat_number
最终函数返回的就是每个基因对,在不同最佳匹配数下的赋值结果。
1 | 0 1 homo1 homo2 homo3 homo4 homo5 |
然后block_position函数, 会用 for k in block[1]的循环提取每个共线性区块中每个基因对的homo值,然后用 df = pd.DataFrame(blk_homo) 和 homo = df.mean().values求均值。
1 | def block_position(self, collinearity, blast, gff1, gff2, ks): |
最终得到的homo1的homo5,是不同最佳匹配基因数下计算的值。如果共线性的点大部分为红色,那么该值接近于1;如果共线性的点大部分为蓝色,那么该值接近于0;如果共线性的点大部分为灰色,那么该值接近于-1。也就是我们可以根据最初的点图中的颜色来确定将来筛选不同WGD事件所产生共线性区块。
这也就是为什么homoN可以作为共线性片段的筛选标准。