第十四章 ArchR的足迹分析
转录因子(Transcripts factor, TF)足迹分析使得我们能够预测特定位点中TF的精确结合位置。这是因为该位置被TF结合避免了转座酶的切割,而TF结合位点的邻近位置处于开放状态。
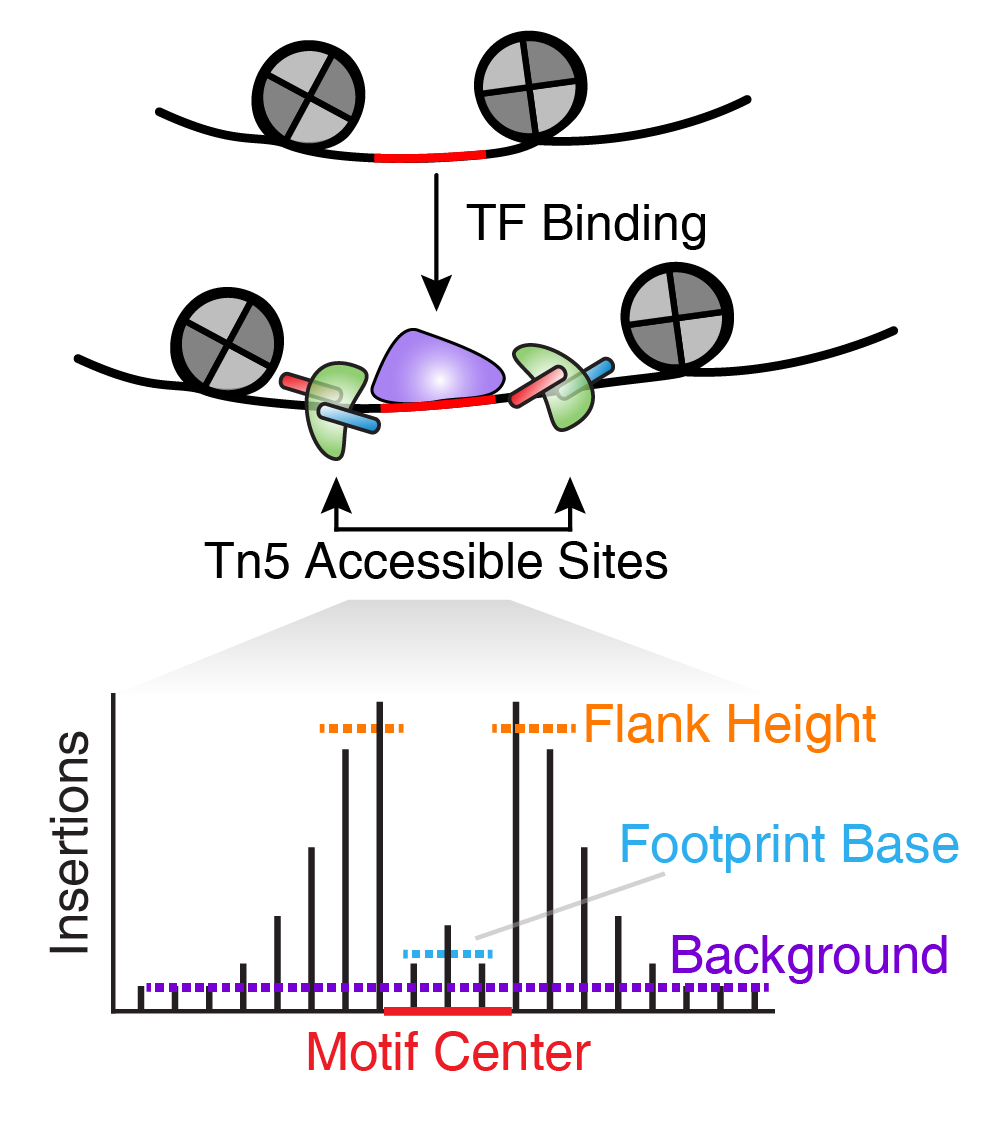
理想情况下,TF足迹分析需要在单个位置上分析从而确定TF的准确结合位置。但实际上,这需要非常高的测序深度,甚至超过混池ATAC-seq或者scATAC-seq的所有数据。为了解决这个问题,我们可以把和待预测的TF结合相关的Tn5插入位置进行合并。例如,我们可以提取所有包含CTCF motif的peak,制作一个全基因组的CTCF的聚合TF足迹。
为了保证足迹的可靠性,我们需要确保能够可靠的预测出目标TF所对应的结合位点。ArchR使用自带的addMotifAnnotations()函数对peak区域进行搜索,寻找能够匹配的DNA序列。考虑到motif的简并性,无法保证每个motif都有足够的peak。添加到ArchRProject的motif注释以二值矩阵表示(0=无motif, 1=有motif)。一旦你有了这些motif注释,ArchR使用getFootPrints()函数分析足迹,它以一个ArchRProject对象和一个GenomicRanges对象(记录motif的位置)作为输入。可以使用getPositions()函数从ArchRProject中提取这些位置。之后足迹可以使用plotFootprints()函数可视化。
或许更重要的是,ArchR的足迹分析能够抵消已知的Tn5插入序列偏好性。ArchR使用一个hexmer位置频率矩阵和一个目标Tn5插入位置上的k-mer频率矩阵来实现该功能。
最终,该流程输出考虑到Tn5插入偏好性的足迹图。
ArchR支持motif足迹分析和用户提供特征的足迹分析,在后续都会讨论。
14.1 motif足迹分析
由于教程用到的数据集比较小,因此利用该数据集得到足迹并不是那么的清晰。使用更大的数据会得到较小变异的足迹。
在分析足迹时,我们要先获取和motif相关的所有位置。这可以通过getPositions函数完成。该函数有一个可选参数, name,用于传入peakAnnotation对象中我们想要获取位置的变量名。如果name=NULL, 那么ArchR会使用peakAnnotation槽(slot)的第一个条目(entry)。在下面的例子中,我们没有指定name, ArchR使用的第一个条目为CIS-BP motifs.
1 | motifPositions <- getPositions(projHeme5) |
这会创建一个GRangesList对象,每个TF motif以不同的GRanges对象进行区分。
1 | motifPositions |
我们提取部分感兴趣的TF motifs用于展示。在我们提取”EBF1”会附带”SREBF1”, 因此我们需要用%ni%显式将其过滤。%ni%函数是R自带函数%in%的相反函数。
1 | motifs <- c("GATA1", "CEBPA", "EBF1", "IRF4", "TBX21", "PAX5") |
为了准确找到TF足迹,我们需要大量的reads。因此,细胞需要进行分组生成拟混池ATAC-seq谱才能用于TF足迹分析。这些拟混池谱之前在peak鉴定时就已经保存为分组覆盖文件。 如果没有在ArchRProject添加分组覆盖信息,则运行如下命令
1 | projHeme5 <- addGroupCoverages(ArchRProj = projHeme5, groupBy = "Clusters2") |
在计算分组覆盖度后,我们可以为之前getFootprints()挑选的一组标记motif计算足迹。即便ArchR已经优化了足迹分析流程,我们也建议先对一部分motif分析足迹,而不是直接分析所有motif。 我们通过positions参数来选择motif。
1 | seFoot <- getFootprints( |
当我们获取了这些足迹,我们可以使用plotFootprints()函数进行展示。该函数能够同时以多种方式对足迹进行标准化。下一节,我们会讨论标准化和实际的足迹图。
14.2 Tn5偏好的足迹标准化
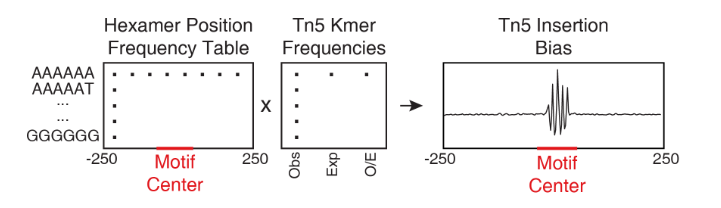
使用ATAC-seq数据分析TF足迹的一大挑战就是Tn5转座酶的插入序列偏好性,这会导致TF足迹的错误分类。为了降低Tn5插入偏好性的影响,ArchR识别每个Tn5插入位置附近的k-mer序列(k由用户提供,默认是6).
对于该项分析,ArchR为每个拟混池识别单碱基分辨率的Tn5插入位点,将这些1-bp位点调整为k-bp窗口(-k/2和+(k/2-1)bp),然后使用Biostrings包中的oligonucleotidefrequency(w=k, simplify.as="collapse")函数创建k-mer频率表。然后,ArchR使用与BSgenome相关的基因组文件,以相同的函数计算出全基因组范围预期的k-mers。
为了计算拟混池足迹的插入偏差,ArchR创建了一个k-mer频率矩阵,该矩阵表示为从motif中心到窗口+/-N bp(用户定义,默认为250 bp)的所有可能k-mer。然后,遍历每个motif位点,ArchR将定位的k-mer填充到k-mer频率矩阵中。然后在全基因组范围内计算每个motif位置。利用样本的k-mer频率表,ArchR可以通过将k-mer位置频率表乘以观察/期望 Tn5 k-mer频率来计算预期的Tn5插入。
以上所有这些发生在plotFootprints()函数中。
14.2.1 减去Tn5偏好
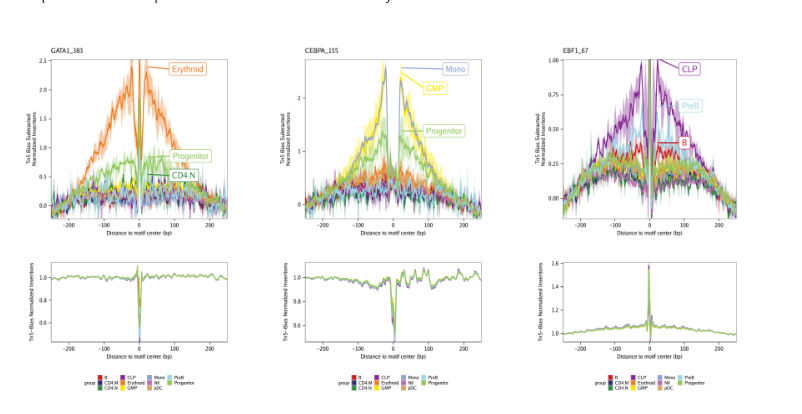
一个标准化方式就是从足迹信号中减去Tn5偏好。该标准化方法通过设置plotFootprints()的normMethod = "Subtract"实现
1 | plotFootprints( |
默认,这些图保存在ArchRProject的outputDirectory。如果你需要绘制所有motif, 可以将其返回为ggplot2对象,需要注意这个ggplot对象会非常大。下面是一个从motif足迹中减去Tn5偏好信号的结果
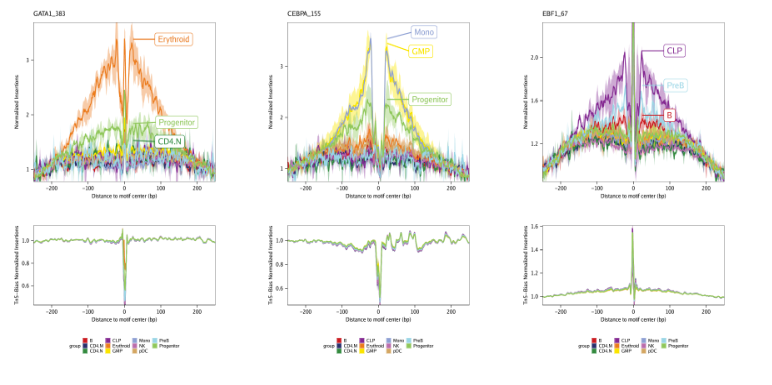
14.2.2 除以Tn5偏好
第二种标准化方法就是将足迹除以Tn5偏好信号。该标准化方法通过设置plotFootprints()的normMethod = "Divide"实现
1 | plotFootprints( |
下面是一个从motif足迹中除以Tn5偏好信号的结果
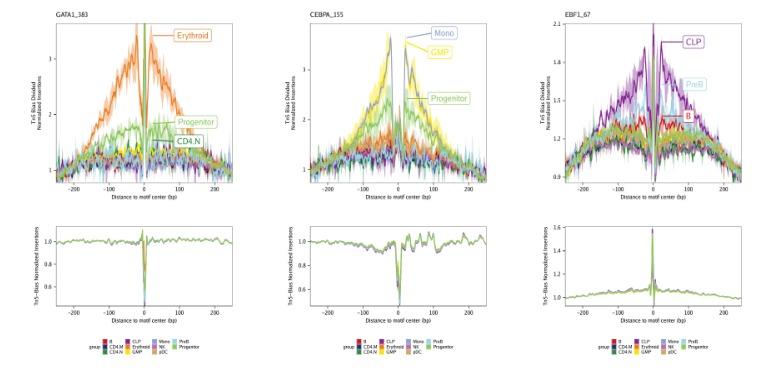
14.2.3 无Tn5偏好标准化的足迹
尽管我们高度推荐将足迹根据Tn5序列插入偏好性进行标准化,当然你可以通过设置plotFootprints()的normMethod = "None"来省去标准化。
1 | plotFootprints( |
14.3 特征足迹
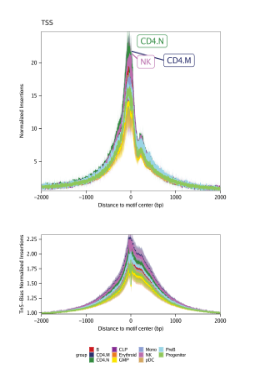
除了motif足迹分析,ArchR还允许用户分析任意定义的特征数据集。为了对功能进行说明,我们将会使用plotFootprints()函数创建TSS插入谱(在之前数据质量控制一节中引入)。一个TSS插入谱本质上就是特殊的足迹。
我们在之前小节讨论过,足迹会用到来源于拟混池重复的分组覆盖文件。我们在之前peak鉴定时创建过这些文件。如果你没有在ArchRProject加入分组覆盖信息, 那么需要运行如下代码
1 | projHeme5 <- addGroupCoverages(ArchRProj = projHeme5, groupBy = "Clusters2") |
我们接着创建一个没有经过Tn5偏好性校正的TSS插入谱。和之前分析一个主要不同是,我们设置了flank=2000, 将TSS向前向后分别延伸2000 bp.
1 | seTSS <- getFootprints( |
我们接着用plotFootprints()对每个细胞分组绘制TSS插入谱。
1 | plotFootprints( |