第15章 使用ArchR进行整合分析
ArchR的一个优势能够整合多种水平的信息从而提供新的洞见。我们可以只用ATAC-seq数据进行分析,如识别peak之间的共开放性来预测调控相互作用,或整合scRNA-seq数据,如通过peak-基因的连锁分析预测增性子活性。无论是哪种情况,ArchR都可以很容易地从scATAC seq数据中获得更深入的见解。
15.1 创建细胞低重叠聚集
ArchR能方便许多特征间相关性的分析。在这些相关分析中,使用稀疏的单细胞数据进行这些计算会导致大量的噪声。为规避这一挑战,我们采用了一种由Cicero引入的方法,在这些分析之前创建单细胞的低重叠聚集。我们过滤与任何其他聚集重叠超过80%的聚集以减少偏差。同时为提升该方法的速度,我们开发了一个优化的迭代重叠检查流程,借由”Rcpp”包通过C++实现了快速特征相关性运算。在ArchR中,这些优化方法被用于计算peak共开放性、peak-基因连锁和其他连锁分析。这些低重叠聚集运算都是在内部完成,为清楚起见,我们先在这里对其介绍。
15.2 ArchR共开放分析
共开放是多个单细胞种的两个peak之间开放性的相关性。换句话说,当peak A在某个单细胞中是开放状态时,peak B通常也是开放状态。下图可以直观地说明了这一概念,说明增强子E3通常与启动子P是共开放的。
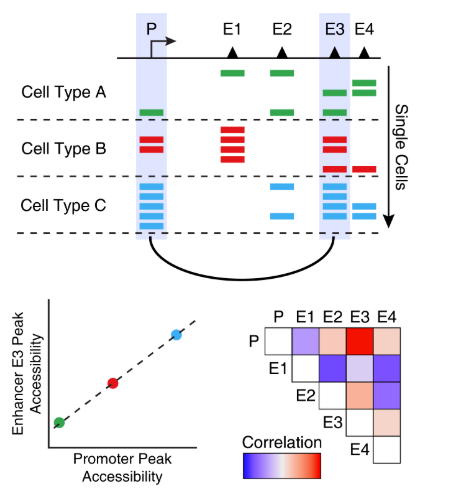
有一点需要注意,共开放分析找到的peak通常都是细胞类型特异的peak。这是因为这些peak在一种细胞类型中都是开放的,而另一种细胞类型通常都是关闭的。虽然这些peak之间有很强的相关性,但是不意味着这些peak之间存在调控关系。
在ArchR中,我们使用addCoAccessibility()函数计算共开放性,计算结束得到的开放性信息保存在ArchRProject中
1 | projHeme5 <- addCoAccessibility( |
使用getCoAccessibility()函数可以从ArchRProject对象中提取共开放性信息,当设置returnLoops=FALSE时会返回一个DataFrame对象。
1 | cA <- getCoAccessibility( |
该DataFrame对象包含几个重要的信息。queryHits和subjectHits列记录的是两个存在相关性的peak的索引。correlation则是两个peak之间的开放状态相关的数值。
1 | cA |
这个共开放DataFrame还有一个元数据成员,包含一个相关peak的GRanges对象。上面提到的queryHits和subjectHits的索引适用于这个GRanges对象。
1 | metadata(cA)[[1]] |
如果我们设置retureLoops=TRUE, 那么getCoAccessibility()会以loop track的形式返回共开放数据。在这个GRanges对象中,IRanges的起始和结束对应的是每个存在互作关系中两个共开放peak的位置。resolution参数设置这些loop的碱基对分辨率。当resolution=1时,它会输出连接每个peak中心的loop.
1 | cA <- getCoAccessibility( |
我们比较下GRanges和DataFrame这两个对象
1 | cA[[1]] |
如果我们将loops的分辨率调整为resolution = 1000, 这能够避免绘制过多的共开放互作事件。从下面的输出的GRanges对象中,我们可以看到条目少了很多
1 | cA <- getCoAccessibility( |
同样的,如果我们进一步降低分辨率resolution = 10000,我们会得到更少共开放交互事件。
1 | cA <- getCoAccessibility( |
15.2.1 在browser track中绘制共开放
当我们在ArchRProject里增加共开放信息后,我们可以用这个信息在基因组浏览器里绘制loop track。具体做法就是在plotBrowserTrack()函数中设置loops参数。我们这里以getCoAccessibility()默认参数输出结果为例,即corCutOff = 0.5, resolution = 1000, returnLoops = TRUE
1 | markerGenes <- c( |
最后用grid.draw函数绘制结果,通过$选择特定的标记基因。
1 | grid::grid.newpage() |
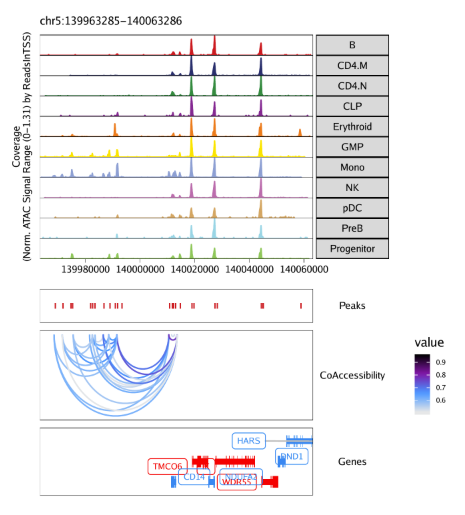
使用plotPDF()保存可编辑的矢量图
1 | plotPDF(plotList = p, |
15.3 ArchR Peak2GeneLinkages分析
和共开放分析类似,ArchR也能分析所谓的”peak-to-gene”关联, 也就是分析peak和基因的相关性。共开放分析和peak-to-gene关联分析的主要不同在于,共开放分析只需要用到ATAC-seq数据,寻找的是peak之间的共开放关系,而peak-to-gene关联分析则会整合scRNA-seq数据,寻找peak开放状态和基因表达量的关系。这其实是相似问题的两种方法。只不过因为peak-to-gene关联分析使用的是scATAC-seq和scRNA-seq数据,我们会认为这个关联更能反应基因调控关系。
在ArchR中,我们使用addPeak2GeneLinks()函数鉴定peak-to-gene的关联。
1 | projHeme5 <- addPeak2GeneLinks( |
我们以上一节类似方式提取这些peak-to-gene的关联信息,只不过这里用的是getPeak2GeneLinks()函数。和之前一样,用户可以设置相关性阈值和关联之间的分辨率。
1 | p2g <- getPeak2GeneLinks( |
当returnLoops=FALSE, 该函数返回一个DataFrame对象,和之前getCoAccessibility()返回的DataFrame相似。主要的不同在于scATAC-seq的索引 peak存放在idxATAC列,而scRNA-seq基因的索引是存放在idxRNA列中
1 | p2g |
peak-to-gene关联DataFrame对象同样也有一个GRanges对象记录对应peak的元信息。上面提到的idxATAC索引能用于GRanges对象中。
1 | metadata(p2g)[[1]] |
如果们设置returnLoop=TRUE,那么getPeak2GeneLinks()会返回一个记录loop track的GRanges对象,链接peak和基因。和之前共开放分析一样,IRanges对象里记录着关联着的peak和基因的位置。当resolution=1, 这连接的是peak的中心和基因的单碱基TSS。
1 | p2g <- getPeak2GeneLinks( |
我们可以将loops的分辨率调整为resolution = 1000, 输出结果主要用于在基因组浏览器中绘制这些连接。因为相同的基因对应的peak过多时,可视化效果会很差。
1 | p2g <- getPeak2GeneLinks( |
进一步降低分辨率,也会降低peak-to-gene连接的总数。
1 | p2g <- getPeak2GeneLinks( |
15.3.1 在browser track中绘制peak-to-gene连接
我们可以采用之前共开放分析流程中的相同方法,使用plotBrowserTrack()函数在基因组浏览器上绘制peak-to-gene 连接。
1 | markerGenes <- c( |
用grid.draw函数绘制结果,通过$选择特定的标记基因。
1 | grid::grid.newpage() |
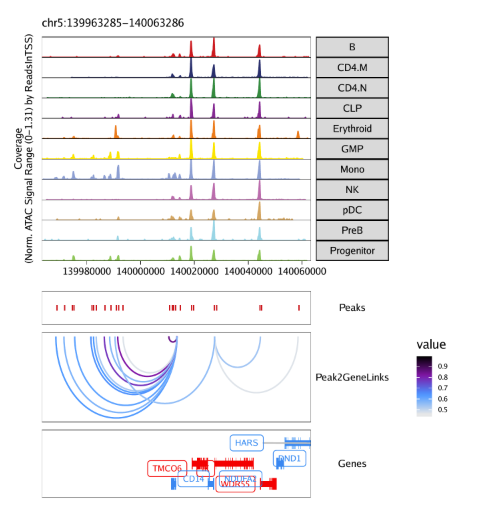
使用plotPDF()保存可编辑的矢量图
1 | plotPDF(plotList = p, |
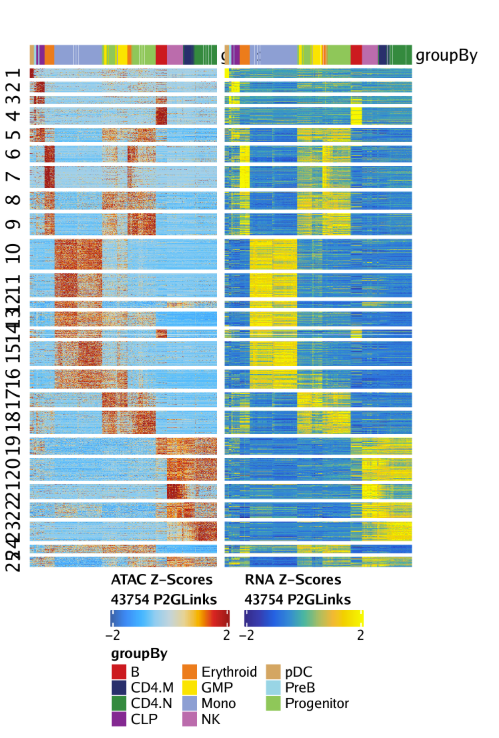
15.3.2 绘制Peak-to-gene连接热图
我们还可以通过绘制peak-to-gene热图的方式展示我们的peak-to-gene连接。热图分为两个部分,一个是scATAC-seq数据,一个是scRNA-seq数据。绘图使用plotPeak2GeneHeatmap()函数
1 | p <- plotPeak2GeneHeatmap(ArchRProj = projHeme5, groupBy = "Clusters2") |
热图的行是k-means聚类结果,其中k由我们进行设置,默认是25.
1 | p |
15.4 正向TF-调控因子鉴定
ATAC-seq还可以无偏好的鉴定TF,这些TF所对应的染色质区域(存在该TF结合的motif DNA序列 )表现出很大改变。此外,从聚集的位置权重矩阵(Position Weight Matrices, PWM)中也能看到这些TF家族在其集合位点上有相似的特征(例如 GATA因子)。

这种motif的相似性使得识别特定的TF充满了挑战性,这些TF可能在其预测的结合位点驱动染色质开放状态发生改变。为了规避这一挑战,我们之前用ATAC-seq和RNA-seq来鉴定TF,这些TF的基因表达和对应motif的开放状态呈正相关。我们将这些TF命名为”正向调节因子”。然而,这种分析依赖于匹配的基因表达数据,而这些数据并不是所有实验中都有。ArchR可以识别其推断基因得分与其chromVAR TF偏差z-score相关的TF来克服这种依赖性。实现方式为,ArchR利用低重叠细胞聚集将TF motif的chromVAR deviation z-scroe和TF基因的基因活跃得分相关联。当ArchR整合scRNA-seq时,TF的基因表达可以代替推测的基因活性评分。
15.4.1 第一步: 鉴定偏离TF motif
鉴定正向TF调控因子的第一步就是鉴定存在偏离的TF motif。我们在之前的章节进行过该分析,为所有的motif创建了一个MotifMatrix,记录着chromVAR deviations 和deviation z-scores。使用getGroupSE()函数可以获取基于聚类平均后的数据,返回的是一个SummarizedExperiment.
1 | seGroupMotif <- getGroupSE(ArchRProj = projHeme5, useMatrix = "MotifMatrix", groupBy = "Clusters2") |
因为SummarizedExperiment对象来自于MotifMatrix, 所以它有两个seqnames, “deviation”和”z”,对应chromVAR里原始的偏离值和偏离值的z-scores.
1 | seGroupMotif |
我们可以从SummarizedExperiment中只提取deviation z-scores.
1 | seZ <- seGroupMotif[rowData(seGroupMotif)$seqnames=="z",] |
然后,我们计算该z-score在所有聚类中最大变化值。根据motif在不同聚类中观察到的差异程度,我们可以对其分层。
1 | rowData(seZ)$maxDelta <- lapply(seq_len(ncol(seZ)), function(x){ |
15.4.2 第二步: 鉴定相关的TF motif和TF基因得分/表达值
接着我们使用correlateMatrices()函数来获取motif开放状态矩阵和基因活跃矩阵(基因得分或基因表达量),在这里就是GeneScoreMatrix和MotifMatrix. 它们将用于鉴定一类motif开放状态和其自身基因活跃相关的TF。正如之前所提到,我们使用许多低重叠细胞聚集在低维空间里(对应reducedDims参数)来计算相关性。
1 | corGSM_MM <- correlateMatrices( |
该函数返回一个DataFrame对象,记录着GeneScoreMatrix和MotifMatrix,以及它们在低重叠细胞聚类中的相关性。
1 | corGSM_MM |
同样的分析还可以使用GeneIntegrationMatrix, 用于替代GeneScoreMatrix
1 | corGIM_MM <- correlateMatrices( |
15.4.3 第三步: 在相关性DataFrame中添加极大偏差值
对于每个相关性分析,我们使用第一步计算的极大偏差值(maximum delta )来注释每个motif
1 | corGSM_MM$maxDelta <- rowData(seZ)[match(corGSM_MM$MotifMatrix_name, rowData(seZ)$name), "maxDelta"] |
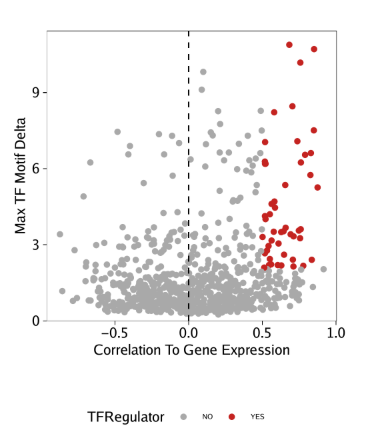
15.4.4 第四步: 鉴定正向TF调控因子
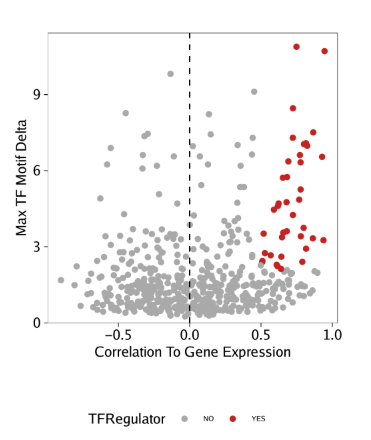
我们可以利用所有这些信息来识别正向TF调控因子。在下面的例子中,正向调控因子的标准为
- motif和基因得分(或基因表达)之间的相关性大于0.5
- 调整后的p值小于0.01
- deviation z-score的最大组间差异位于前四分位。
我们应用这些过滤标准,并且做一些文本调整来分离TF名字。
1 | corGSM_MM <- corGSM_MM[order(abs(corGSM_MM$cor), decreasing = TRUE), ] |
在从基因得分和motif deviation z-scores鉴定到正向TF调控因子后,我们可以在点图中突出展示
1 | p <- ggplot(data.frame(corGSM_MM), aes(cor, maxDelta, color = TFRegulator)) + |
使用相同的分析策略处理GeneIntegrationMatrix
1 | corGIM_MM <- corGIM_MM[order(abs(corGIM_MM$cor), decreasing = TRUE), ] |