尽管目前已测序的物种已经很多了,但是对于一些动辄几个G的复杂基因组,还没有某个课题组有那么大的经费去测序,所以仍旧缺少高质量的完整基因组,那么这个时候一个高质量的转录组还是能够凑合用的。
二代测序的组装结果只能是差强人意,最好的结果就是不要组装,直把原模原样的转录组给你是最好的。PacBio Iso-Seq 做的就是这件事情。只不过Iso-Seq测得是转录本,由于有些基因存在可变剪切现象,所以所有将一个基因的所有转录本放在一起看,搞出一个尽量完整的编码区。
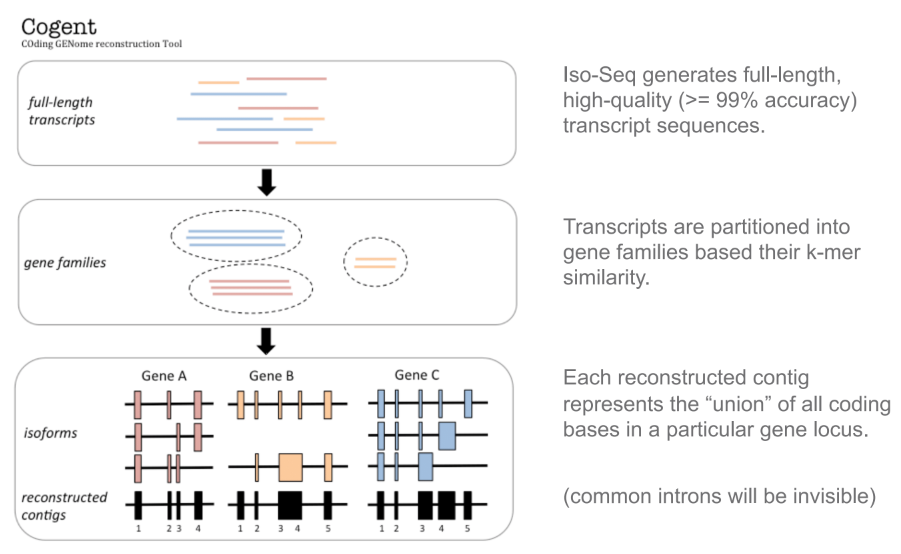
Cogent能使用高质量的全长转录组测序数据对基因组编码区进行重建,示意图如下:
软件安装
虽然说软件可以直接通过conda进行安装,但是根据官方文档的流程,感觉还是很麻烦
下面操作默认你安装好了miniconda3, 且miniconda3的位置在家目录下
安装方法如下(更新与2020/7/30)
1 | conda create -y -n cogent python=3.7 |
经过上面一波操作,请用下面这行命令验证是否安装成功
1 | run_mash.py --version |
此外还需要安装另外两个软件,分别是Minimap2和Mash
1 | conda install minimap2 |
对待大数据集,你还需要安装Cupcake
1 | conda activate cogent |
关于这个Cupcake, 如果python3环境出现报错,就新建一个Python2环境进行安装。
创建伪基因组
让我们先下载测试所用的数据集,
1 | mkdir test |
第一步: 从数据集中搜索同一个基因簇(gene family)的序列
这一步分为超过20,000 条序列的大数据集和低于20,000条序列这两种情况, 虽然无论那种情况,在这里我们都只用刚下载的测试数据集
小数据集
第一步:从输入中计算k-mer谱和配对距离
1 | run_mash.py -k 30 --cpus=12 test_human.fa |
你一定要保证你的输入是fasta格式,因为该工具目前无法自动判断输入是否是fasta格式,所以当你提供诡异的输入时,它会报错,然后继续在后台折腾。
上面这行命令做的事情是:
- 将输入的fasta文件进行拆分,你可以用
--chunk_size指定每个分块的大小 - 对于每个分块计算k-mer谱
- 对于每个配对的分块,计算k-mer相似度(距离)
- 将分块合并成单个输出文件
第二步:处理距离文件,创建基因簇
1 | process_kmer_to_graph.py test_human.fa test_human.fa.s1000k30.dist test_human human |
这里的test_human是输出文件夹,human表示输出文件名前缀。此外如果你有输入文件中每个转录亚型的丰度,那么你可以用-c参数指定该文件。
这一步会得到输出日志test_human.partition.txt,以及test_human下有每个基因family的次文件夹,文件里包含着每个基因簇的相似序列。对于不属于任何基因family的序列,会在日志文件种专门说明,这里是human.partition.txt
大数据集
如果是超过20,000点大数据集,分析就需要用到Minimap2和Cpucake了。分为如下几个步骤:
- 使用minimap2对数据进行粗分组
- 对于每个分组,使用上面提到的精细的寻找family工具
- 最后将每个分组的结果进行合并
第一步:使用minimap进行分组
run_preCluster.py要求输入文件名为isoseq_flnc.fasta, 所以需要先进行软连接
1 | ln -s test_human.fa isoseq_flnc.fasta |
为每个分组构建基因簇寻找命令
1 | generate_batch_cmd_for_Cogent_family_finding.py --cpus=12 --cmd_filename=cmd preCluster.cluster_info.csv preCluster_out test_human |
得到的是 cmd 文件,这个cmd可以直接用bash cmd运行,也可以投递到任务调度系统。
最后将结果进行合并
1 | printf "Partition\tSize\tMembers\n" > final.partition.txt |
第二步:重建编码基因组
上一步得到每个基因簇, 可以分别重构编码基因组,所用的命令是reconstruct_contig.py
1 | 使用方法: reconstruct_contig.py [-h] [-k KMER_SIZE] |
如果你手头有一个质量不是很高的基因组,可以使用-G GENOME_FASTA_MMI和-S SPECIES_NAME提供参考基因组的信息。毕竟有一些内含子是所有转录本都缺少的,提供基因组信息,可以补充这部分缺失。
由于有多个基因簇,所以还需要批量运行命令 reconstruct_contig.py
1 | generate_batch_cmd_for_Cogent_reconstruction.py test_human > batch_recont.sh |
第三步: 创建伪基因组
首先获取未分配的序列, 这里用到get_seqs_from_list.py脚本来自于Cupcake, 你需要将cDNA_Cupcake/sequence添加到的环境变量PATH中。
1 | tail -n 1 human.partition.txt | tr ',' '\n' > unassigned.list |
这里的测试数据集里并没有未分配的序列,所以上面这一步可省去。
然后将未分配的序列和Cogent contigs进行合并
1 | mkdir collected |
最后,将冗余的转录亚型进行合并。 这一步我们需要用Minimap2或者GMAP将Iso-seq分许得到的hq_isoforms.fasta回帖到我们的伪基因组上。 关于参数选择,请阅读Best practice for aligning Iso Seq to reference genome: minimap2, GMAP, STAR, BLAT
1 | ln -s ../test_human.fa hq_isoforms.fasta |
然后可以根据collapse tutorial from Cupcake将冗余的转录亚型合并
1 | sort -k 3,3 -k 4,4n hq_isoforms.fasta.sam > hq_isoforms.fasta.sorted.sam |
由于这里没有每个转录亚型的丰度文件cluster_report.csv,所以下面的命令不用运行, 最终结果就是hq_isoforms.fasta.no5merge.collapsed.rep.fa
1 | get_abundance_post_collapse.py hq_isoforms.fasta.no5merge.collapsed cluster_report.csv |
如果运行了上面这行代码,那么最终文件就应是hq_isoforms.fasta.no5merge.collapsed.filtered.*