第11章: 使用ArchR鉴定标记Peak
在讨论基因得分(gene score)这一章中,我们已经介绍了标记特征的鉴定。相同的函数getMakerFeatures()也能够用于从ArchRProject任意矩阵中鉴定标记特征。所谓的标记特征指的是相对于其他细胞分组唯一的特征。这些特征能帮助我们理解类群或者细胞类型特异的生物学现象。在这一章中,我们会讨论如何使用该功能鉴定标记peak。
11.1: 使用ArchR鉴定标记Peak
通常而言,我们想知道哪些peak是某个聚类或者某一些聚类所特有的。在ArchR中,这可以通过设置addMarkFeatures()函数的useMatrix="PeakMatrix"来实现(无需监督)。
首先,我们需要再看一眼projHeme5中有哪些细胞类型,以及它们的相对比例
1 | table(projHeme5$Cluster2) |
现在,让我们调用getMarkerFeatures参数,并设置useMatrix="PeakMatrix". 此外,为了降低不同细胞组之间的数据质量对结果的影响,我们可以设置bias参数,其中bias = c("TSSEnrichment", "log10(nFrags)")就是用来避免TSS富集和每个细胞的fragment数对结果的影响。
1 | markersPeaks <- getMarkerFeatures( |
getMarkerFeatures()函数返回一个SummarizedExperiment对象,该对象包含一些不同的assays
1 | markerPeaks |
接着,我们可以用getMarkers函数从输出的SummarizedExperiment对象中提取我们感兴趣的部分。默认情况下,它会返回一个包含多个DataFrame的列表,不同的DataFrame表示来自不同的细胞分组。
1 | markerList <- getMarkers(markersPeaks, cutOff = "FDR <= 0.01 & Log2FC >= 1") |
如果我们对特定的一个细胞分组感兴趣,我们可以用$进行提取。
1 | markerList$Erythroid |
除了返回一个包含多个DataFrame的列表外,我们还可以用getMarkers()返回一个GRangesList,只要设置returnGR=TRUE即可。
1 | markerList <- getMarkers(markersPeaks, cutOff = "FDR <= 0.01 & Log2FC >= 1", returnGR = TRUE) |
这个GRangesList同样可以用$提取特定细胞组的结果,返回的是一个GRanges对象
1 | markerList$Erythroid |
11.2 在ArchR中绘制Marker Peaks
ArchR提供了许多绘图函数用于getMarkerfeatuers()返回的SummarizedExperiment对象的可视化。
11.2.1 Marker Peak Heatmap
markerHeatmap能以热图的形式展示标记Marker Peak(或者其他getMarkerFeatures()输出的特征)
1 | heatmapPeaks <- markerHeatmap( |
使用draw函数绘制结果
1 | plotPDF(heatmapPeaks, name = "Peak-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE) |
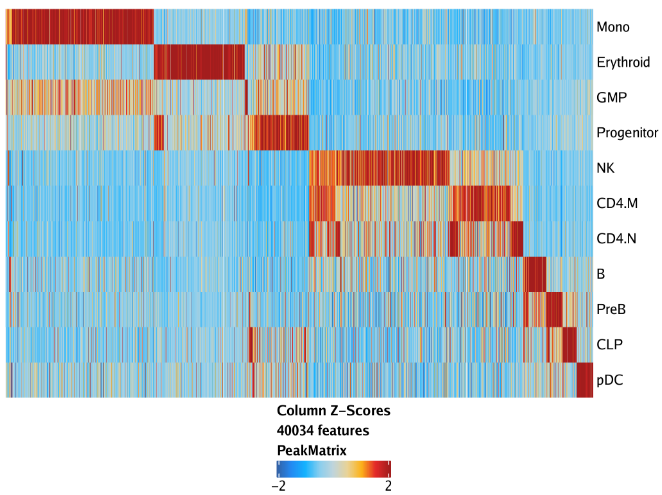
plotFDF()函数能够以可编辑的矢量版本保存图片
1 | plotPDF(heatmapPeaks, name = "Peak-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme5, addDOC = FALSE) |
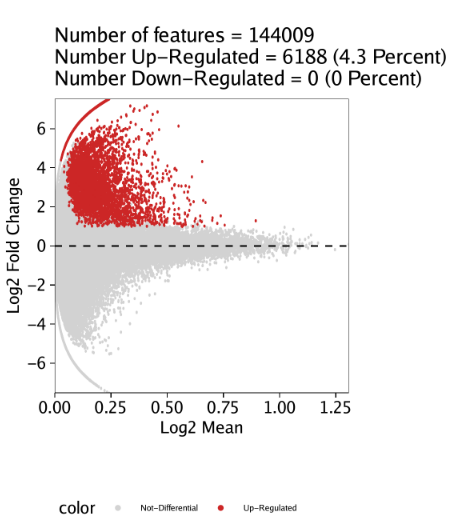
11.2.2 Marker Peak MA和火山图
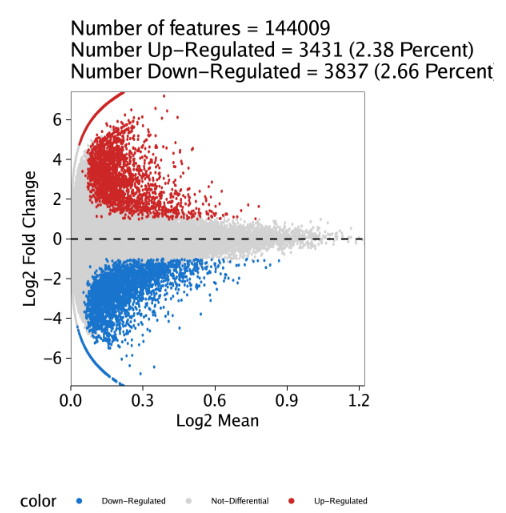
除了绘制热图,我们也可以为每个细胞分组绘制MA或者火山图(volcano)。这些图可以用markerPlot()函数绘制。对于MA图,需要设置参数plotAs="MA". 我们以”Erythroid”细胞分组为例,设置参数name = "Erythroid"
1 | pma <- markerPlot(seMarker = markersPeaks, name = "Erythroid", cutOff = "FDR <= 0.1 & Log2FC >= 1", plotAs = "MA") |
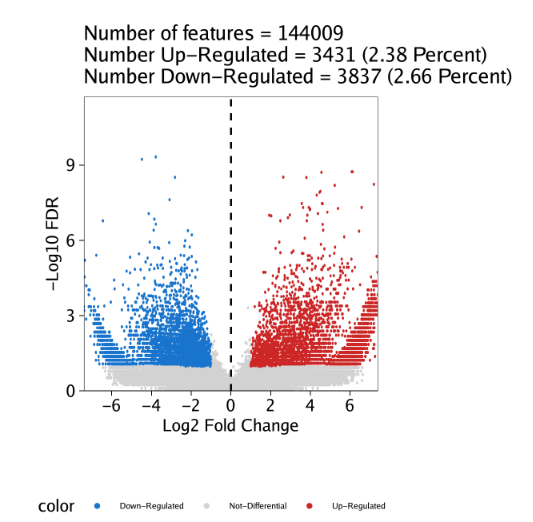
同样的,只要设置plotAs="Volcano"就可以绘制火山图
1 | pv <- markerPlot(seMarker = markersPeaks, name = "Erythroid", cutOff = "FDR <= 0.1 & Log2FC >= 1", plotAs = "Volcano") |
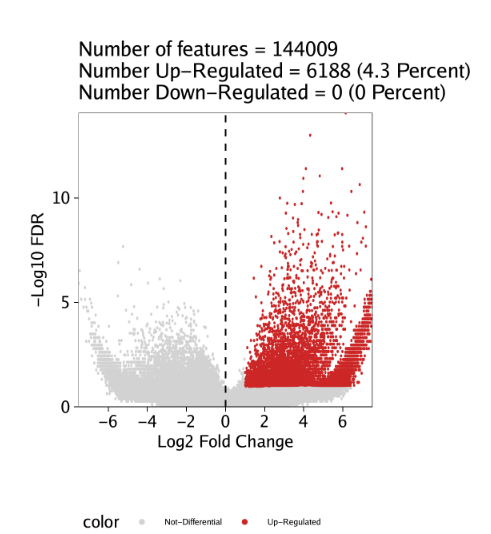
plotFDF()函数能够以可编辑的矢量版本保存图片。
1 | plotPDF(pma, pv, name = "Erythroid-Markers-MA-Volcano", width = 5, height = 5, ArchRProj = projHeme5, addDOC = FALSE) |
11.2.3 Browser Tracks的Marker Peak
此外,我们在基因组浏览器上检查这些peak区域,只需要为plotBrowserTrack()函数的features参数传入 相应的peak区间。这会额外在我们的ArchR track图的下方以BED形式展示marker peak区域。
1 | p <- plotBrowserTrack( |
我们使用grid::grid.draw()绘制结果
1 | grid::grid.newpage() |
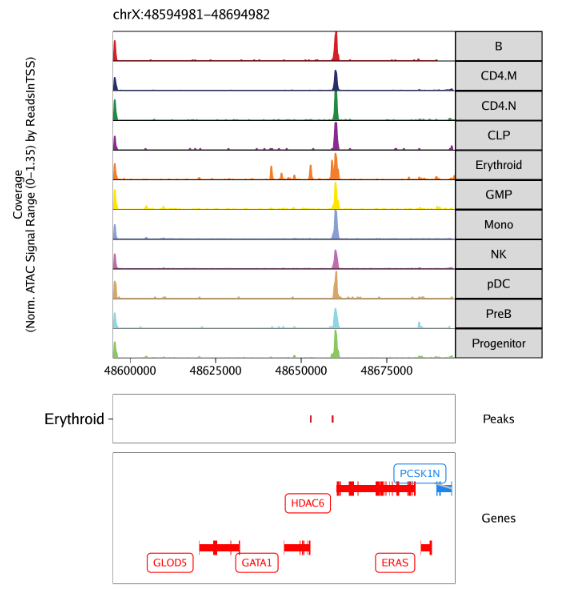
plotFDF()函数能够以可编辑的矢量版本保存图片。
1 | plotPDF(p, name = "Plot-Tracks-With-Features", width = 5, height = 5, ArchRProj = projHeme5, addDOC = FALSE) |
11.3 组间配对检验
标记特征鉴定是一种特别的差异表达检验。此外,使用相同的getMarkerFeatures()函数也能实现标准化的差异分析。我们只需要设置useGroup为一组细胞,然后设置bgdGroup为另一组细胞即可。这就可以对给定两组进行差异分析。在这些差异分析中,在useGroups比较高的peak的倍数变化值是正数,在bgdGroups比较高的peak则是由负的倍数变化值。
这里,我们对”Erythroid”与”Progenitor”细胞组进行配对检验。
1 | markerTest <- getMarkerFeatures( |
使用markerPlot()函数可以绘制MA或者火山图。MA图需要设置plotAs="MA"
1 | pma <- markerPlot(seMarker = markerTest, name = "Erythroid", cutOff = "FDR <= 0.1 & abs(Log2FC) >= 1", plotAs = "MA") |
火山图需要设置plotAs="Volcano"
1 | pv <- markerPlot(seMarker = markerTest, name = "Erythroid", cutOff = "FDR <= 0.1 & abs(Log2FC) >= 1", plotAs = "Volcano") |
plotFDF()函数能够以可编辑的矢量版本保存图片。
1 | plotPDF(pma, pv, name = "Erythroid-vs-Progenitor-Markers-MA-Volcano", width = 5, height = 5, ArchRProj = projHeme5, addDOC = FALSE) |
在后续章节中,我们还会进行差异分析,因为会在我们的差异开放的peak中搜索富集的motif。