第7章: ArchR的基因得分和标记基因
尽管ArchR能够可靠地对细胞进行聚类,但它并不能先验(a priori)地知道每个聚类所代表的细胞类型。通常这个任务也只能靠人工注释,毕竟每个项目都不太一样。
为了实现细胞类型注释,我们会用到已知的细胞类型特异的标记基因。在scATAC-seq数据中,这些基因的表达量其实是根据染色质开放数据估计的基因得分(gene score)。所谓的基因得分,本质上就是用基因附近的调控元件去预测基因的表达量。ArchR的亮点在于,它允许用户提供复杂的自定义的距离加权开放性模型去计算这些基因得分。
7.1 在ArchR中计算基因得分
我们在文章中测试了超过50种不同的基因得分模型,并找到一种在大部分测试情况下都拥有良好表现的模型。这个模型目前是ArchR的默认设置,它包括三个主要部分
- 在整个基因主体内(gene body)的开放部分都对基因得分有贡献
- 对于潜在的远距调控元件,我们使用一个指数加权函数计算得分
- 我们加入了基因边界(gene boundary)用于减少无关调控元件对基因得分的贡献
那么,ArchR到底是怎么计算基因得分的呢?我建议先看图,再看文字描述。图中标注了两个TSS,分别表示两个基因的转录起始位点(TSS),其中红色标识的基因是目标基因,我们需要根据其附近的开放情况来预测它的表达量。

对于每条染色体,ArchR都会构建一个分块矩阵(tile matrix),块的大小由用户进行定义,在定义时计算(默认是500 bp),然后分析用户定义的另一个基因分窗(默认是基因两边100kb)和上一个分块矩阵的重叠部分。接着计算每个分块的起始位置或结束位置相对于基因主体(可额外添加上下游延伸)或基因起始位置的距离。我们发现基因表达量最好的预测者是基因区的局部开放性,包括启动子和基因主体。对于之前提到的远距调控元件,为了能正确地处理给定基因的远距离开放状态,ArchR只筛选了在基因分窗内但不跨到另一个基因区的分块。这个过滤标准既能利用远端调控元件提高基因表达量的预测准确性,同时也避免了无关开放状态的影响(例如,邻近基因启动子的开放状态)。每个分块到基因的距离会根据用户定义的开放模型里的权重进行转换,默认是e(-abs(distance)/5000) + e-1)。
我们还发现基因长度会明显影响总体的基因得分,这是因为如果基因区包括基因主体,当基因越长时,落在基因主体内的开放区也就越多,这些开放区会和最大的基于距离的权重相乘,也就会计算出更高的得分。为了调整基因长度所带来的差异,ArchR以每个基因长度的倒数(1 / gene size)作为权重,然后将该权重线性缩放到1到用户定义的最大值之间(默认是5)。保证了越短的基因对应更大的相对权重,一定程度上避免了长度的影响。
之后每个分块对应的距离权重和基因长度权重会和每个分块内的Tn5 insertion数相乘,接着将基因分窗内的所有分块的得分进行相加,相加时排除邻近基因区的分块。最后计算的总开放度就是基因得分(gene score),接着所有基因的基因得分会按照用户定义的常数进行深度标准化(默认是10,000)。最后,计算的基因得分会被保存在相应的Arrow文件中用作下游分析。
在创建Arrow文件时,如果设置createArrowFiles的参数addGeneScoreMat = TRUE(默认为TRUE),那么生成的Arrow文件中会包含基因得分矩阵。此外,我们也可以在任意时候使用addGeneScoreMatrix()在Arrow文件中加入基因得分矩阵。一旦计算完成,每个细胞都可以在嵌入图中根据基因得分进行展示,方便鉴定不同的细胞类型。之后的章节中会逐一介绍基因得分的应用。
当然并不是所有基因都会有基因得分,尤其是基因密度高的区域里的基因,它们更容易出现问题。因此,最好能在基因组浏览器对所有基因得分都做一遍检查,我们也会在后续章节里做相应介绍。
7.2 鉴定标记特征
除了使用已知的细胞类型相关标记基因用于聚类注释外,ArchR还能无偏地鉴定任何给定细胞分组(如聚类)的标记特征。这些特征包括单不限于peaks, 基于基因得分的基因, 基于chromVAR离差的转录因子motif。这部分工作可通过getMarkerFeatures()函数完成,它的核心参数是useMatrix和groupBy。useMatrix可以是任何的矩阵输入,如果设置为GeneScoreMatrix那么函数就会分析不同细胞类型特异出现的基因。这就能无偏的找到每个聚类的活跃基因,可以用来辅助聚类注释。
正如之前所提到的,getMarkerFeatures()能够接受任何存在Arrow文件里的矩阵作为输入,分析细胞类型的特异特征。例如useMatrix = "TileMatrix"用于鉴定细胞类群的特异基因区间,useMatrix = "PeakMatrix"用于鉴定细胞类群的特异peak。后续章节还会介绍如何使用其他特征类型作为getMarkerFeatures()的输入。
7.2.1 标记特征的鉴定过程
标记特征的鉴定过程取决于每一组细胞对应的偏好-匹配背景细胞的选择。对于所有特征,每个细胞组都会与它自己的背景细胞组进行比较,判断给定的细胞组有更显著的开放性。

选择合适的背景细胞组对标记特征的鉴定至关重要,取决于getMarkerFeatures)()的bias参数所构建的多维空间。对于给定分组的细胞,ArchR会在提供的多维空间中寻找每个细胞分组外的最近邻细胞。这些细胞和给定分组内细胞极其相似,因此被称为偏好-匹配细胞(bias-matched cells)。通过这一方法,即便是在细胞数目比较少的组中,我们都能为每个特征计算稳健的显著性。
ArchR以bias参数中的所有维度作为输入,以分位数对值进行归一化,让每个维度的方差分布在相同的相对标度。举一个简单的例子,如如果bias的输入是TSS和log10(Num Fragments),未经分位数归一化的值如下图所示

这里y轴的相对方差和于x轴的相对方差相比,就显得特别小。如果我们对这些轴进行归一化,将值缩放到0-1之间,x和y的相对方差就近乎一样。注意,这个操作还会明显地改变每个细胞的最近邻(见下图右)。

ArchR会对所有维度进行归一化,在归一化后的多维空间中以欧氏距离寻找最近邻。
7.3 鉴定标记基因
为了根据基因得分来鉴定标记基因,我们需要在调用getMarkerFeatures()时设置useMatrix = "GeneScoreMatrix。此外,我们设置了groupBy="Clusters"告诉ArchR使用cellColData的”Clusters”列对细胞分组,从而得到每个聚类特异的标记基因。
1 | markersGS <- getMarkerFeatures( |
该函数返回一个SummarizedExperiment对象,里面记录着每个标记特征的相关信息。这是ArchR的一个常见输出,是下游数据分析的关键输出格式。SummarizedExperiment对象和矩阵类似,行是感兴趣的特征(例如基因),列表示样本。一个SummarizedExperiment能够记录一个或多个assay矩阵。如果你需要了解更多和SummarizedExperiment相关的内容,建议阅读相关的Bioconductor页面,这里就不深入对这方面内容进行介绍。
我们能够使用getMarkers()函数从SummarizedExperiment对象中提取出一个包含多个DataFrame对象的列表,每个DataFrame对象都对应着一个聚类,记录着该聚类里相关的标记特征。
1 | markerList <- getMarkers(markersGS, cutOff = "FDR <= 0.01 & Log2FC >= 1.25") |
我们使用markerHeatmap()创建含有所有标记特征的可视化热图。通过labelMarkers()在热图中标记部分感兴趣的基因。
1 | markerGenes <- c( |
我们需要用ComplexHeatmap::draw()才能绘制该热图,这是因为heatmapGS实际上是一个热图列表。
1 | ComplexHeatmap::draw(heatmapGS, heatmap_legend_side = "bot", annotation_legend_side = "bot") |

使用plotPDF()函数保存可编辑的矢量版。
1 | plotPDF(heatmapGS, name = "GeneScores-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme2, addDOC = FALSE) |
7.4 在嵌入上可视化标记基因
在之前章节中提到,我们可以在UMAP嵌入上展示每个细胞的基因得分。这可以通过修改plotEmbedding()函数里的colorBy和name参数实现
1 | markerGenes <- c( |
选择绘图列表的其中一个基因进行展示
1 | p$CD14 |

如果是需要绘制所有基因,那么可以使用cowplot将不同的标记基因整合到一张图中
1 | p2 <- lapply(p, function(x){ |

使用plotPDF()函数保存可编辑的矢量版。
1 | plotPDF(plotList = p, |
7.5 使用MAGIC填充标记基因
在上一节中,你可能注意到一些基因得分图变化很大,这是因为scATAC-seq数据太过稀疏。我们使用MAGIC根据邻近细胞填充基因得分对信号进行平滑化处理。我们发现者能够极大程度地提高基因得分的可视化解读性。要执行此操作,我们需要先在我们的ArchRProject中加入填充权重。
1 | projHeme2 <- addImputeWeights(projHeme2) |
这些填充权重会在之后绘制UMAP嵌入图里的基因得分时传入到plotEmbedding()
1 | markerGenes <- c( |
和之前一样,我们可以只画其中一个基因

也可以用cowplot绘制所有的标记基因
1 | #Rearrange for grid plotting |

使用plotPDF()函数保存可编辑的矢量版。
1 | plotPDF(plotList = p, |
7.6 使用ArchRBrowser绘制Track
除了在UMAP上绘制每个细胞的基因得分外,我们还可以在基因组浏览器上浏览这些标记基因的局部染色体开放状态。为了执行该操作,我们使用plotBrowserTrack()函数,它会构建一系列绘图对象,每个对象对应一个标记基因。函数会根据groupBy输入的组别信息在不同track上绘制每组的开放状态。
1 | markerGenes <- c( |
通过选择列表中的给定基因来绘制最终结果
1 | grid::grid.newpage() |

使用plotPDF()函数可以将多幅基因座位对应的图保存在一个PDF文件中。
1 | plotPDF(plotList = p, |
7.7 启动ArchRBrowser
scATAC-seq数据分析一个与生俱来的挑战就是在基因组浏览器上可视化聚类间的染色质开放水平。传统做法是,先对scATAC-seq的fragments进行分组,然后构建一个基因组覆盖度的bigwig文件,最后对track进行标准化后才能实现定量层面上的可视化。终端用户经常用于测序数据可视化的基因组浏览器包括 WashU Epigenome Browser, UCSC Genome Browser, IGV browser。这一过程需要用到多个不同软件,并且一旦细胞分组信息改变,或者增加了更多的样本,就得重新分组输出bigiwig文件,相当耗时。
为了避免重复劳动,ArchR开发了基于Shiny的交互式基因组浏览器,只需要一行ArchRBrowser(ArchRProj)命令就可启动。因为数据都存放在Arrow文件中,所以这个交互式浏览器就能动态的更改细胞分组信息,分辨率以及标准化,实现实时的track水平的可视化。ArchR Genome Browser 同样也能生成高质量的PDF格式输出文件用于发表或分享。此外,浏览器支持用户通过features参数传入GenomicRanges对象用来展示特征,或者通过loop参数传入基因组交互文件( co-accessibility, peak-to-gene linkages, loops from chromatin conformation data)。其中loop的预期格式是GRanges,起点表示其中一个loop的中心,终点表示另一个loop中心。
使用ArchRBrowser()函数启动我们的本地交互基因组浏览器
通过选择”Gene Symbol”,就可以开始浏览了。你可能需要点击”Plot Track”才能强制让你的浏览器刷新
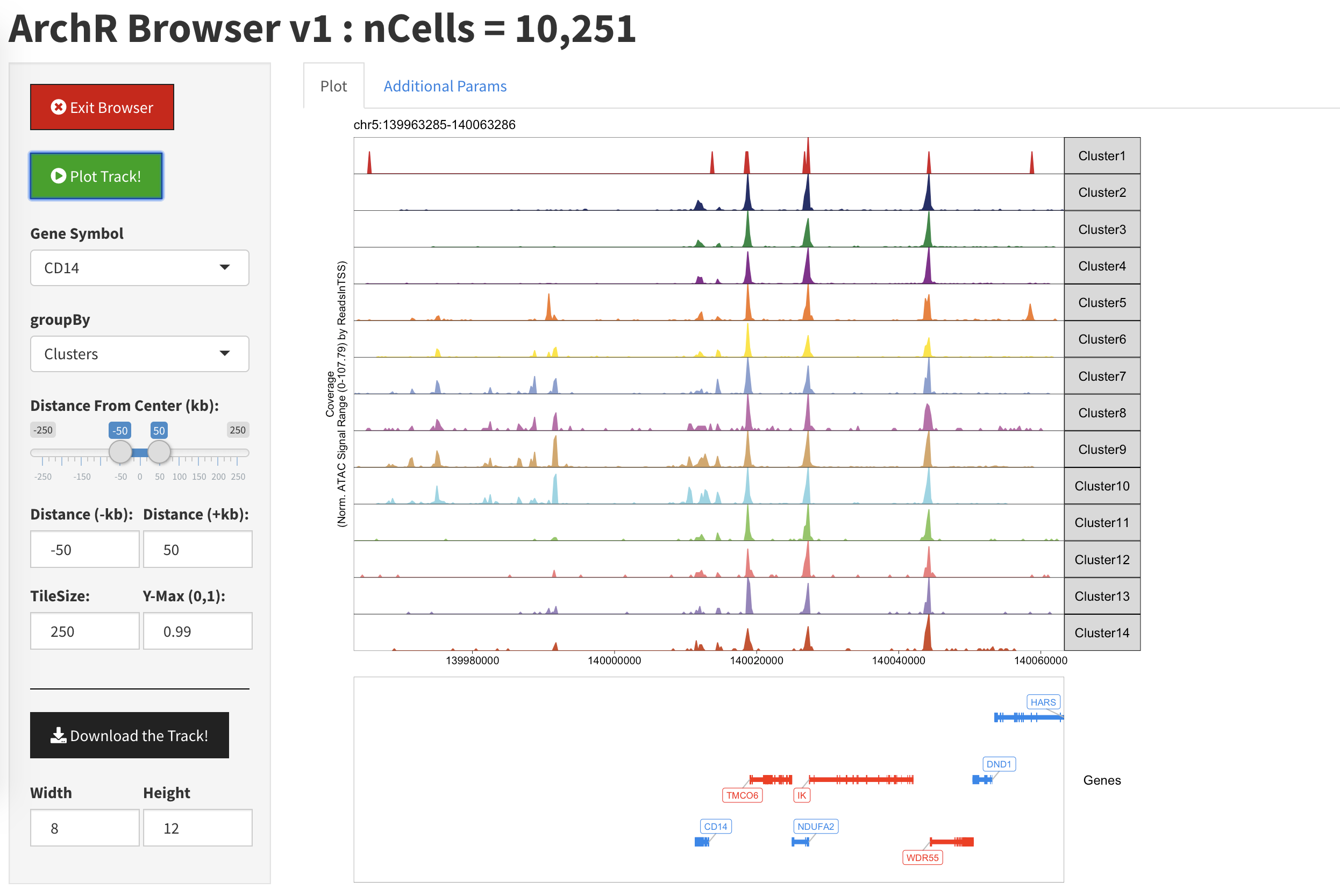
当我们绘制一个基因位点后,我们看到不同track代表的是我们数据的不同聚类。
如果我们点击”Additional Parameters”侧边栏,我们可以选择部分聚类进行展示
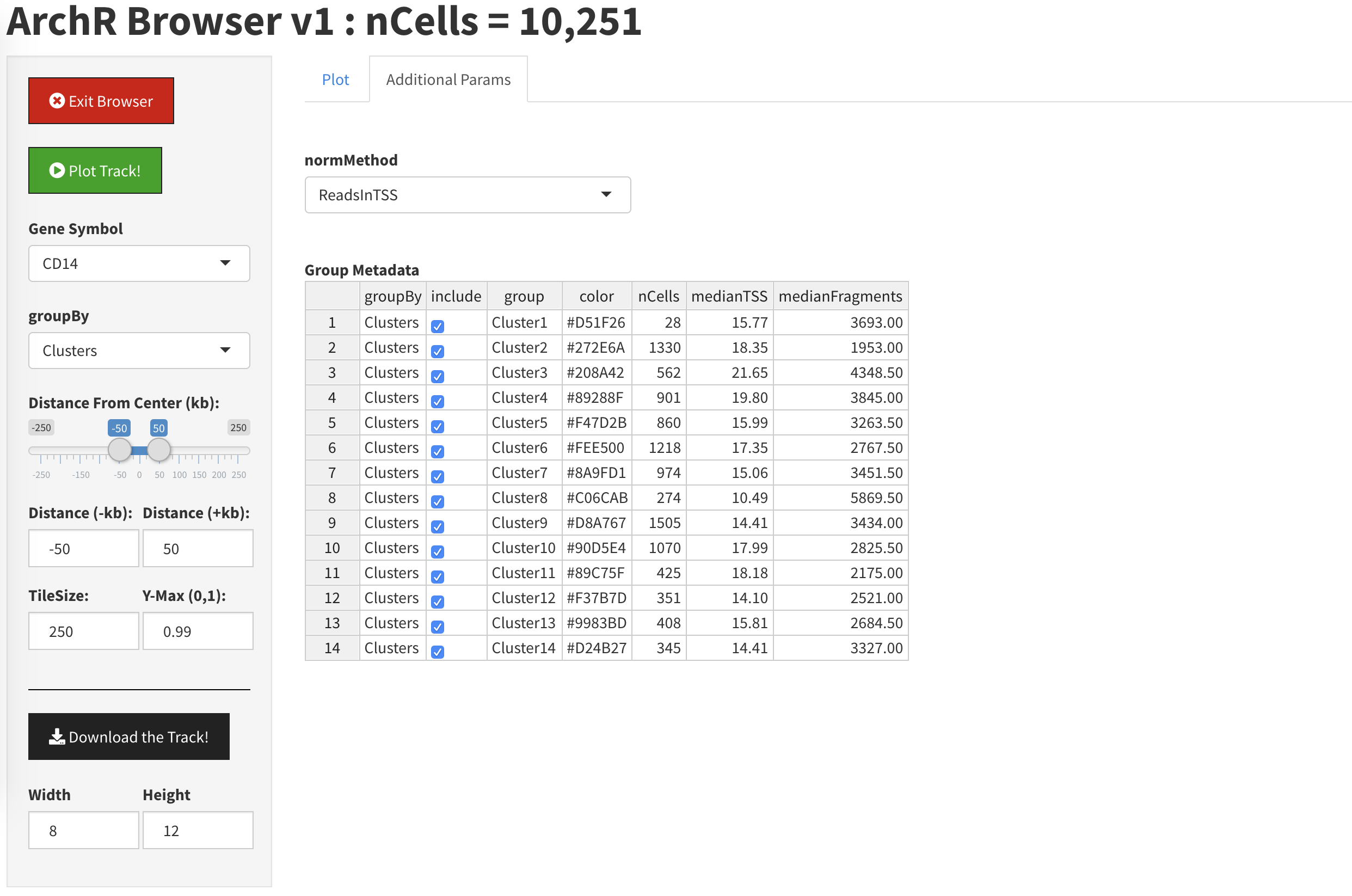
通过反选cluster1, 2, 3,我们就在绘图中移除了它们
当我们返回到”Plot”时,此时Cluster1,2,3就从图片中消失了。同样你也可能需要点击”Plot Track”才能让浏览器刷新结果
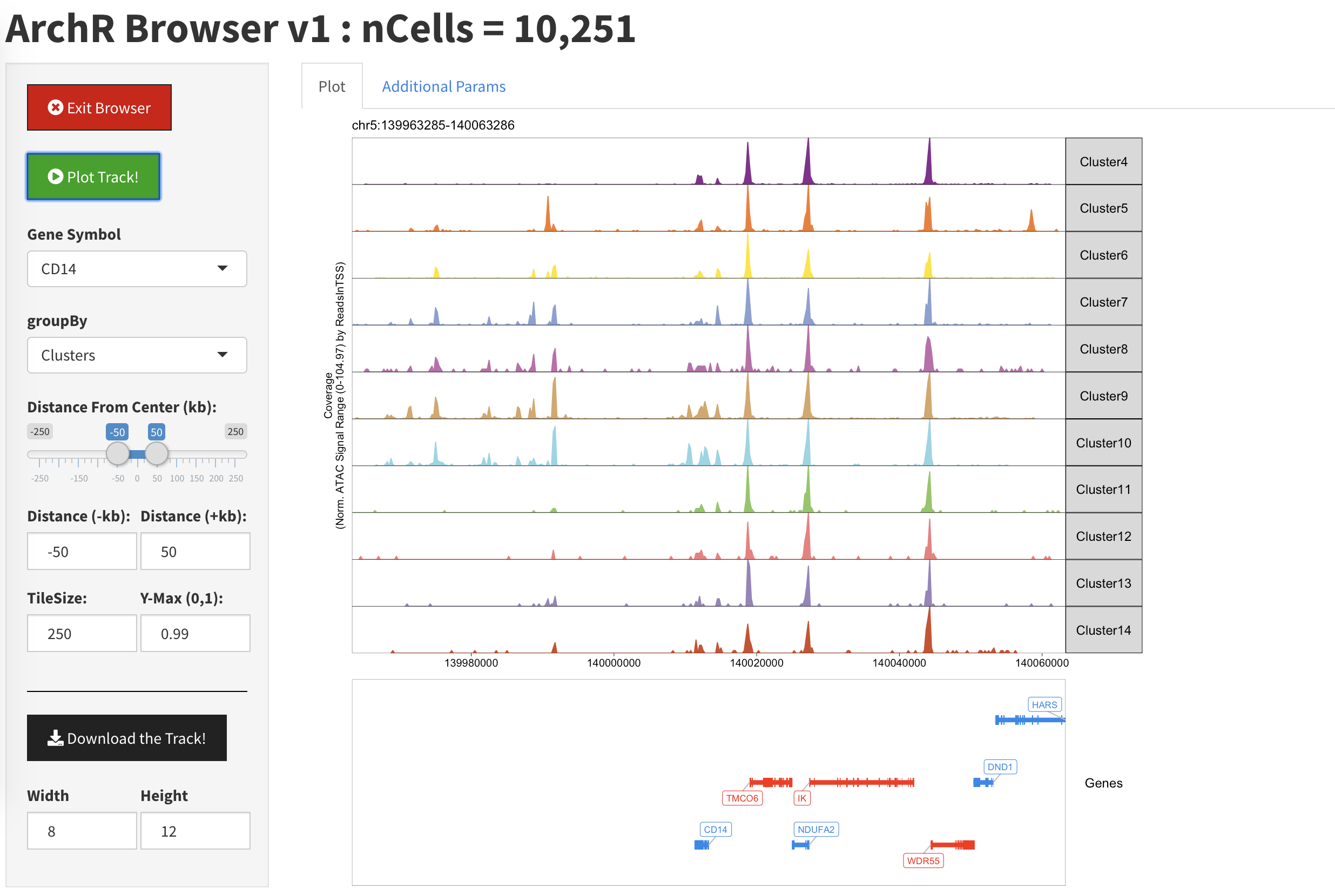
无论在哪个阶段,我们都可以点击”Download The Track”来输出当前的绘图结果。