purge_dups能够根据read深度分析组装中haplotigs和overlaps。相对于另一款purge_haplotigs,它的运行速度更快,而且能够自动确定阈值。
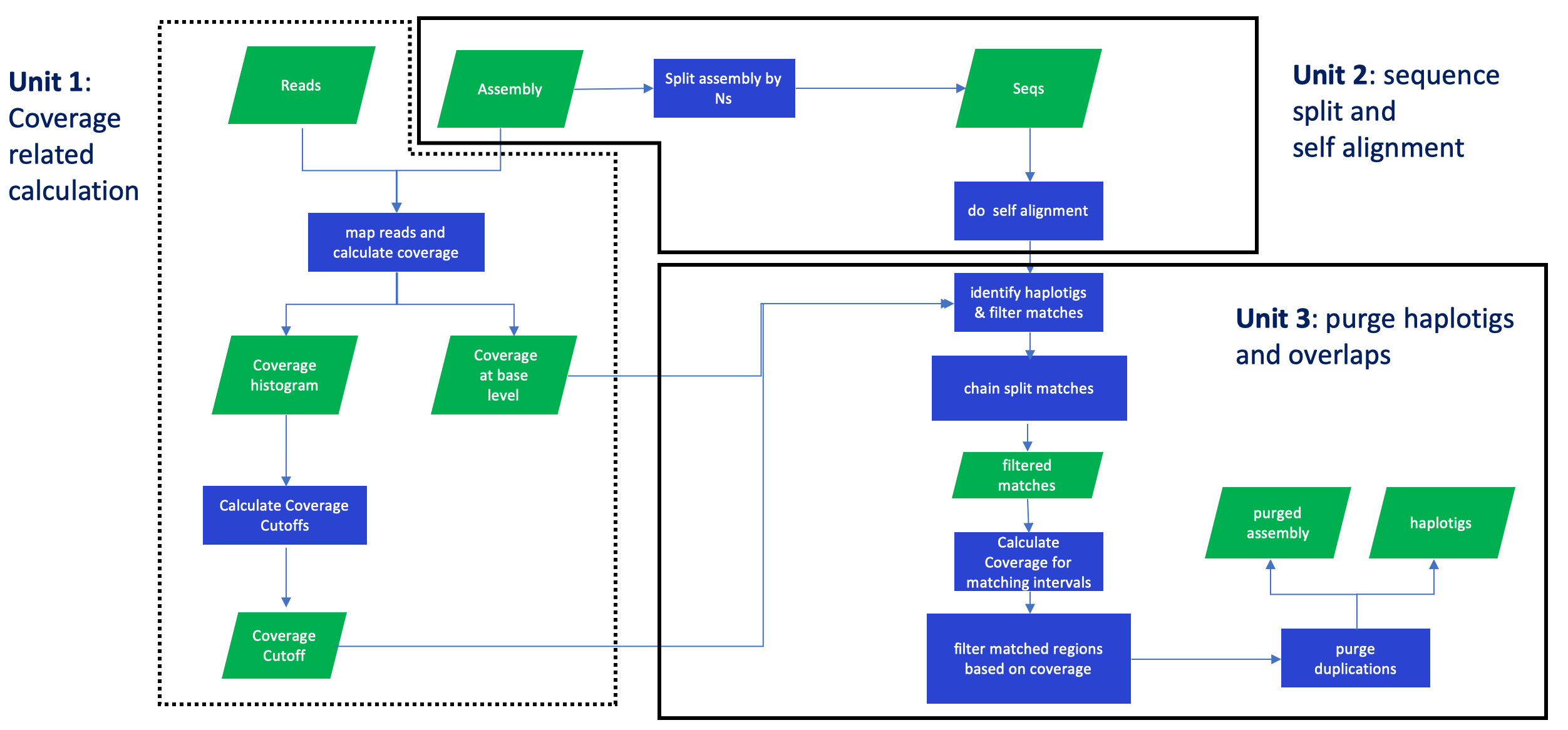
purge_dups分为三个部分,第一部分是将序列回贴到基因组并分析覆盖度确定阈值,第二部分是将组装自我比对,第三部分是利用前两部分得到的信息鉴定到原来序列中的haplotigs和overlaps.
软件安装
尽管可以通过runner来进行程序调用,但是我更喜欢自己写脚本,因此不安装python3和runner
purge_dups是用C语言编写,因此需要通过make来编译
1 | git clone https://github.com/dfguan/purge_dups.git |
脚本在scripts目录下,编译的程序在bin目录下
软件运行
输入文件分为两种,一种是组装序列,一种是测序数据。其中组装序列分为两种情况考虑,一种是类似falcon-unzip输出的primary assembly和alternative assembly,另一种则是单个组装文件。 而测序数据分为二代测序和三代ONT/PacBio测序。这里以单个组装文件输出和三代测序进行介绍,假定这两个输入文件分别命名为asm和reads.
第一步: 根据覆盖度计算分界点(cutoff)
1 | # gzip可以替换成pigz, 进行多线程压缩 |
如果是二代测序,可以用bwa mem进行比对,然后用bin/ngscstat统计输出的bam文件覆盖度信息,然后用bin/calcuts计算分界点。
第二步: 将assembly从N处进行打断,如果assembly中没有N那就不会被打断,然后使用minimap2进行contig的自我比对。
1 | # Split an assembly |
这一步可以和前一步同时运行,两者互不影响。
第三步: 根据每个碱基的覆盖度以及组装的自我比对结果来对contig进行分类。
1 | # purge haplotigs and overlap |
dups.bed里的第四列就是每个contig的分类信息,分为”JUNK”, “HIGHCOV”, “HAPLOTIG”, “PRIMARY”, “REPEAT”, “OVLP” 这6类,其中只有
purge_dups可以先以默认参数进行运行,如果结果不理想,可以调整如下参数
-f默认是.8, 根据80%区域的覆盖度来对contig进行分类。例如80%的区域都低于5x,将该序列定义为JUNK。对应源码中的classify_seq函数的min_frac参数-a和-b用来过滤alignment, 对于源码中的flt_by_bm_mm的min_bmf和min_mmf参数-m表示将两个联配衔接时,最低的匹配碱基数-M和-G:分别表示第一轮和第二轮将前后两个联配衔接时最大的空缺大小-E表示 如果合并之后的alignment在contig末尾的前15k内,那么就把alignment延伸至contig末尾-l: 用于控制overlap的大小,该值越小,overlap越多
第四步: 使用get_seqs根据dups.bed从原来的contig中提取主要组装和单倍型。
1 | # get the purged primary and haplotigs sequences from draft assembly |
这里的purged.fa就是最终结果,junk, haplotig和duplication都会在hap.fa中。
可选步骤: 将alternative assembly和输出度hap.fa进行合并,然后运行上面四步,得到的purge.fa就是新的alternative assembly,而输出的hap.fa则是junk或overrepresented序列。
PS: 能不能用来过滤纯合基因组组装的垃圾序列呢?根据我对一个物种的测试,过滤前后的BUSCO值,几乎没有变化,missing rate只提高了0.1%,
1 | # 运行前 |
因此我觉得这种用法是可行的,且Canu的作者也建议用purge_dups处理,参考canu-issues-1717。
另外,作者尚未在ONT和Illumina数据中测试该软件,但是作者认为只需要修改minimap2的-x map-pb为-x map-ont就可以用在ONT数据上。