一句话评价:重复序列注释用EDTA就完事了。
简介
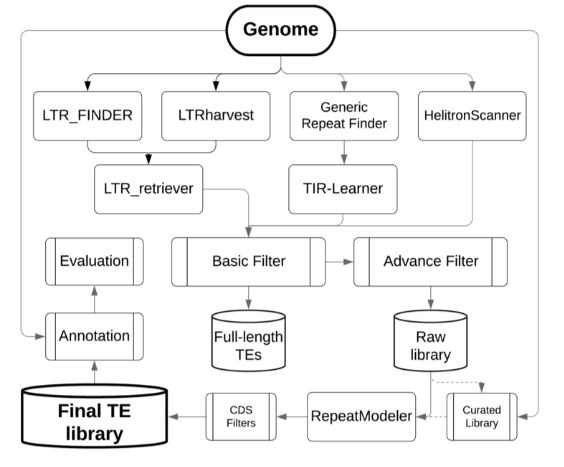
EDTA, 全称是 Extensive de-novo TE Annotator, 一个综合性的流程工具,它整合了目前LTR预测工具结果,TIR预测工具结果,MITE预测工具结果,Helitrons预测工具结果, 从而构建出一高可信,非冗余的TE数据库,用做基因组的注释。流程图如下
安装
如果没有管理员权限,可以用conda进行安装。如果有管理员权限,可以尝试用docker或者singularity进行安装。
PS: 如果之前用过EDTA的话,可以更新一下版本,因为从1.7.0开始,EDTA把intact TE和homology based注释结合在一起,最终产生了很高质量的gff,合并了所有注释;1.7.1版把包含基因的intact也去掉了,进一步过滤。(来自于作者的建议)
conda
使用conda的安装方法如下
1 | conda create -n EDTA |
需要注意的一点是,bioconda建议添加国内镜像站点, 否则可能会下载失败。
singularity
方法1: 使用EDTA上提供的docker镜像,以singularity进行安装
1 | singularity build edta.sif docker://kapeel/edta |
在使用时,有一点需要注意,需要用-B将外部的RepeatMasker的Libraries绑定的Libraries,否则可能会在检查依赖这一步失败。
这里,我参考的是LoReAN的方法
1 | cd ~ # 切换到家目录 |
上面的/home/xzg/是我的家目录,需要根据实际情况进行选择
方法2: 使用 @wangshun1121 构建的docker镜像, 他解决了需要-B进行挂载的问题。
1 | singularity build edta.sif docker://registry.cn-hangzhou.aliyuncs.com/wangshun1121/edta |
实战
我们以拟南芥的第一条染色体为例,进行介绍
1 | # singularity |
这里的参数比较简单,
-genome: 输入的基因组序列-species: 物种名,Rice, Maize和others三个可选-step: 运行步骤,all|filter|final|anno, 根据具体情况选择-t: 线程数,默认是4
此外还有几个参数可以关注下
-cds: 提供已有的CDS序列(不能包括内含子和UTR),用于过滤.这个值也比较重要,建议提供下,否则会降低busco值(来自于作者的推荐)-sensitive: 是否用RepeatModeler分析剩下的TE,默认是0,也就是不要。RepeatModeler运行时间比较久,量力而信。-anno: 是否在构建TE文库后进行全基因组预测,默认是0.-evalues 1: 默认是0,需要同时设置-anno 1才能使用。建议加上,它能够查看注释质量,是非常不错的功能哦(来自于作者的推荐)
运行结束之后,会在当前目录下留下运行时的中间文件,保证你程序中断之后,能够断点续跑xxx.EDTA.raw
xxx.EDTA.combine
xxx.EDTA.final
以及你关注的xxx.EDTA.TElib.fa, 这就是最终的TE文库。
需要注意的是,在实际运行的时候,你不能单条染色体的运行,这不是程序设计的目的,我们这里用一条染色体仅仅是为了演示,测试程序能否顺利运行。
而在实际项目中,一定要用所有的染色体或者scaffold
可能问题
我在使用EDTA时,就遇到了两个问题。一个是singularity的EDTA直接使用时无法通过依赖检测,解决方法已经在安装部分提过,这里不在赘述。
另一个问题我在”Identify TIR candidates from scratch”这一步出现下面的报错
1 | what(): terminate called after throwing an instance of 'Resource temporarily unavailable std::system_error' |
我对这个报错进行了分析,找到了对应代码,即sh $TIR_Learner -g $genome -s $species -t $threads -l $maxint. 用实际内容替换变量后,即下面这行代码
1 | sh /EDTA/bin/TIR-Learner2.4/TIR-Learner2.4.sh -g chr.fa -s others -t 20 -l 5000 |
更具体一点,可以将问题定位到脚本的Module 3, Step 3: Get dataset
1 | genomeFile=/data/xzg_data/1800_assembly/annotation/repeatAnnotation/chr.fa #基因组文件的实际路径 |
将线程数设置为1后,该代码顺利跑通。进一步,我定位getDataset.py的出问题的地方实际是predict函数。当然接着执行后续的代码,发现改动这一参数并不影响下面代码的运行。
1 | echo "Module 3, Step 4: Check TIR/TSD" |
我发现predict函数涉及到了Python的多进程调用,最终在偶然间找到问题真正所在,即Linux系统对用户的资源限制,可以通过ulimit -a查看。
最终我通过设置ulimit -u 9000,提高允许运行的总程序数,将问题解决。
参考资料
- EDTA官方文档
- RMblast的问题
- 一个关于fork资源不够的解决过程
- Ou, S., Su, W., Liao, Y., Chougule, K., Agda, J.R.A., Hellinga, A.J., Lugo, C.S.B., Elliott, T.A., Ware, D., Peterson, T., et al. (2019). Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biology 20, 275.