BioNano以cmap格式存放光学图谱,为了评估基因组的组装质量或者了解光学图谱中冗余情况(高杂合基因组组装结果偏大),我们就需要进行cmap之间的比较。
CMAP间比对
Solve套件提供了runCharacterize.py脚本封装了RefAligner,用于进行CMAP之间的比对。
1 | python2.7 runCharacterize.py \ |
需要注意的是-p和-a参数的设置。-p是Pipeline的文件位置,比如说我的Solve安装在/opt/biosoft/Solve3.4_06042019a,那么参数设置为 -p /opt/biosoft/Solve3.4_06042019a/Pipeline/06042019。 而-a则是要在/opt/biosoft/Solve3.4_06042019a/RefAligner/8949.9232rel/目录下选择合适的xml文件。比如你的CMAP是Irys平台,那么你可以考虑用optArguments_nonhaplotype_irys.xml.
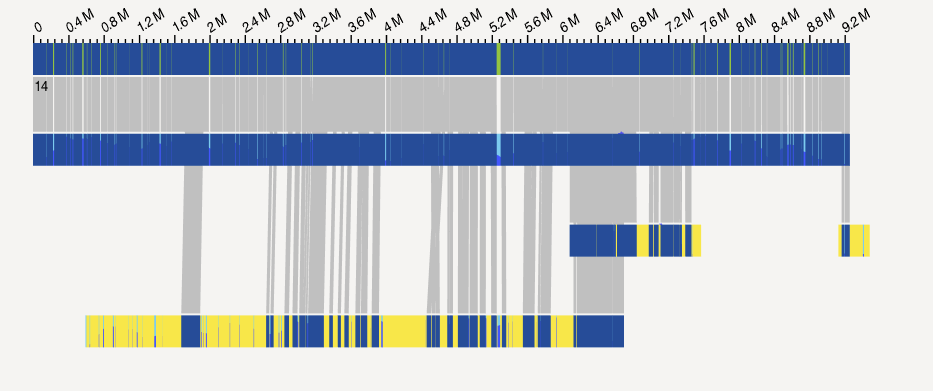
以最新发表的辣椒的光学图谱为例,该物种有比较高的杂合度,组装结果偏大,我们可以通过自比对来寻找冗余区域,
1 | # 下载CMAP |
最终会在当前文件下生成一个alignRef文件夹,其中结果是q.cmap,r.cmap和xmap的文件可以用于上传到BioNano Access上进行展示。下图就是一个冗余实例,可以把图中较短的图谱删掉
基因组回帖
为了将基因组回帖到CMAP上,需要先将基因组的fasta格式转成CMAP格式,参数如下
1 | perl fa2cmap_multi_color.pl -i 输入FASTA -e 酶1 通道1 [酶2 通道2] |
其中一个最重要的参数就是酶切类型。例如我需要将序列回帖到用Nt.BspQI酶切组装的光学图谱上,因此运行参数如下
1 | perl /opt/biosoft/Solve3.4_06042019a/HybridScaffold/06042019/scripts/fa2cmap_multi_color.pl -i athaliana.fa -e BspQI 1 |
最后的athaliana_BSPQI_0kb_0labels.cmap就是模拟酶切的CMAP序列。
之后将模拟酶切的结果回帖到实际的CMAP
1 | python /opt/biosoft/Solve3.4_06042019a/Pipeline/06042019/runCharacterize.py \ |
最终会在当前文件下生成一个alignRef文件夹,其中结果是q.cmap,r.cmap和xmap的文件.