SALSA最初是在BMC genomics上发表,应该是当时最早提出利用Hi-C对contig进行纠错的软件,随后3D-DNA引入了这一策略。而最近升级之后的SALSA又在PLoS Computational Biology发表,可能是目前第一个用组装软件输出的GFA信息的工具。
使用Hi-C辅助组装,有以下的可能问题
- 原先的contig存在错误,scaffold引入了之前的错误
- 输入contig过短时,朝向错误引起inversion
SALSA2的优势
- 整合了assembly graph
- 能够对原始的contig进行纠错
它的工作流程如下:
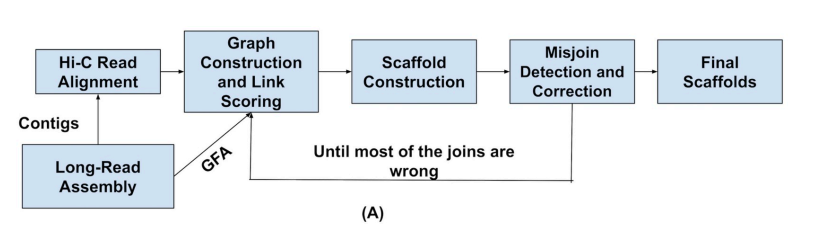
SALSA2从PacBio/ONT的组装结果开始,建议提供记录模棱两可重建位置的GFA文件。Hi-C数据回贴到contig上,contig根据Hi-C的覆盖度分析潜在的组装错误,然后被打断。之后根据GFA和Hi-C信息构建hybrid scaffold graph,最后scaffold根据该graph进行重建。
SALSA提出了一种新的染色体划分方式,也就是检查每次迭代后是否存在错误的合并(mis-join),如果发现大部分的join都是错误的,也意味着目前scaffold划分是最佳的,就可以停止了。
A mis-join detection step is performed after each iteration to check if any of the joins made during this round are incorrect. Incorrect joins are broken and the edges blacklisted during subsequent iterations. This process continues
until the majority of joins made in the prior iteration are incorrect. This provides a natural stopping condition, when accurate Hi-C links have been exhausted.
软件安装
SALSA2的依赖环境如下:
- Python 2.7
- Networkx version < 1.2
- BOOST libraries
通过编译的方法进行安装
1 | git clone https://github.com/marbl/SALSA.git |
软件使用
软件要求以下几个输入文件:
- 三代组装结果: contigs.fasta
- Hi-C比对后BAM: alignment.bam
- (可选) GFA文件: contigs_graph.gfa
第一步: 将Hi-C回贴到组装结果中,这一步主要用bowtie2/bwa比对原始的fastq文件,之后对Hi-C的比对结果进行一些预处理。目前已经有一些流程工具可以实现这一步
- https://github.com/ArimaGenomics
- https://github.com/nservant/HiC-Pro
- https://github.com/Zhong-Lab-UCSD/HiCtool
- https://github.com/XiaoTaoWang/HiC_pipeline
第二步: 将BAM转成BED,并按照Read Name进行排序。假设上一步的输出是alignment.bam
1 | bamToBed -i alignment.bam > alignment.bed |
如果经HiC-Pro处理的数据,那么在hic_results下有一个.allValidPairs, 可以通过awk进行转换成所需要的bed
1 | awk 'BEGIN{OFS="\t"} {print $2,$3,$3+50,$1"/1","60",$4} {print $5,$6,$6+50,$1"/2","60",$7}' input.allValidPairs > alignment.bed |
注意: 关于这个alignment.bed文件,经过我的测试,只需要按照Read Names排序即可,并不需要保证两行两行的配对,SALSA会预先处理。
第三步: 构建contig长度文件
1 | samtools faidx contigs.fasta |
第四步: 根据需求挑选命令运行. 注意-e {Your Enzyme}需要根据实际情况修改,如果是MboI,就写-e GATC.
需求1: 不需要纠错,最基本的组装
1 | python run_pipeline.py -a contigs.fasta -l contigs.fasta.fai -b alignment.bed -e {Your Enzyme} -o scaffolds |
需求2: 使用Hi-C对输入assembly进行纠错(加入参数-m yes)
1 | python run_pipeline.py -a contigs.fasta -l contigs.fasta.fai -b alignment.bed -e {Your Enzyme} -o scaffolds -m yes |
断裂位点位置存放在输出的input_breaks
注意: 关于纠错,建议阅读下assembly error detection isn’t much efficient on my data,这里面在讨论了,Hi-C信号选择对结果的影响。我也在实践中发现使用过滤后的信号会影响最终的breaks位点的数目。
需求3: 使用GFA文件辅助组装(加入了参数-g contigs_graph.gfa)
1 | python run_pipeline.py -a contigs.fasta -l contigs.fasta.fai -b alignment.bed -e {Your Enzyme} -o scaffolds -m yes -g contigs_graph.gfa |
需求4: 使用更加准的unitigs和unitigs GFA. 组装软件,例如Canu会输出xxx.unitigs.fasta和xxx.unitigs.gfa, 里面的序列相对于contig更短,但是错误更少,在某些情况下更适合用于scaffold。
1 | python run_pipeline.py -a unitigs.fasta -l unitigs.fasta.fai -b alignment.bed -e {Your Enzyme} -o scaffolds -m yes -g unitigs_graph.gfa |
最终在-o指定的目录下输出结果,其中scaffolds_FINAL.fasta和scaffolds_FINAL.agp是两个最主要的结果。
如果你因为用了unitigs导致scaffolds中gap太多,那么SALSA可以根据contigs.fasta来关闭gap
1 | python stitch.py -c contigs.fasta -u unitigs.fasta -p scaffolds_iteration_x.p -o output_scaffolds.fasta |
其中scaffolds_iteration_x.p里的x取决于你的迭代次数。
假如你需要在Juicebox中可视化结果,SALSA提供了conver.sh用于数据转换。你需要先从https://github.com/aidenlab/juicer/wiki/Download下载Juicer tools jarw.
1 | wget https://s3.amazonaws.com/hicfiles.tc4ga.com/public/juicer/juicer_tools_1.14.08.jar |
然后修改SALSA的convert.sh中的JUICER_JAR为你实际路径,最后运行
1 | bash /path/to/SALSA/convert.sh SALSA_OUTDIR |
输出文件是salsa_scaffolds.hic
SALSA的所有参数说明如下:
- -a/–assembly: 初始的组装结果,header中不能有
: - -l/–length: contigs的长度
- -b/–bed: BAM文件转换后的bed
- -o/–output: 输出文件夹
- -c/–cutoff: 用于scaffold的contig最短长度
- -g/–gfa: GFA文件
- -e/–enzyme: 限制性内切酶
- -i/–iter: 迭代数
- -x/–dup: 存放重复的contig信息
- -s/–exp: 期望的基因组大小
- -m/–clean: 是否检查Mis-assembly
不客观的测评
SALSA这个工具无论是安装还是使用上,都非常的简洁易懂。我在使用过程中就没有遇到过安装错误,并且运行效率高(基于Python和C++)。最终的组装结果可能和文章说的那样,得到相对最优的scaffolds,无法得到染色体级别的输出结果。
Building on this work, SALSA2 does not require manual parameter tuning and is able to utilize all the contact information from the Hi-C data to generate near optimal sized scaffolds permitted by the data using a novel iterative scaffolding method
SALSA的结果的确是比较可靠的,在contig的朝向上出错更少。不过,对于mis-assembly的鉴定,目前SALSA可能有些问题,在我实践中发现有些明显的错误它没有找到。
此外,在我的几个项目实战中,我发现3D-DNA能够得到染色体水平的结果的情况,而SALSA却无法得到相同的结果。或许是输入的HiC信号不同,之后会尝试用Juicer的输出作为SALSA的输入信号,进行比较。
参考资料
- Integrating Hi-C links with assembly graphs for chromosome-scale assembly
- https://github.com/marbl/SALSA
其他我写过的HiC工具