inferCNV用与探索肿瘤单细胞RNA-seq数据,分析其中的体细胞大规模染色体拷贝数变化(copy number alterations, CNA), 例如整条染色体或大片段染色体的增加或丢失(gain or deletions)。工作原理是,以一组”正常”细胞作为参考,分析肿瘤基因组上各个位置的基因表达量强度变化. 通过热图的形式展示每条染色体上的基因相对表达量,相对于正常细胞,肿瘤基因组总会过表达或者低表达。
inferCNV提供了一些过滤参数,通过调整参数来降低噪音,更好的揭示支持CNA的信号。此外inferCNV还包括预测CNA区间的方法以及根据异质性模式定义细胞类群的方法。
软件安装
尽管inferCNV是一个R包,但是在安装inferCNV之前还需要先下载安装JAGS ,好在它有Windows,MacOS和Linux版本,所以inferCNV在各个平台都能用。
Windows和MacOS的JAGS容易安装,而Linux的JAGS需要编译
1 | # 手动安装BLAS和LAPACK不推荐 |
安装R包
1 | install.packages("rjags") |
测试安装
1 | library(infercnv) |
如果没有报错,就说明安装成功。
软件使用
准备输入文件
需要准备3个输入数据
- 单细胞RNA-seq表达量的原始矩阵
- 注释文件,记录肿瘤和正常细胞
- 基因或染色体位置文件
第一个是Genes x Cells的表达矩阵(matrix),行名是基因,列名是细胞编号。
| MGH54_P16_F12 | MGH54_P12_C10 | MGH54_P11_C11 | MGH54_P15_D06 | MGH54_P16_A03 | … | |
|---|---|---|---|---|---|---|
| A2M | 0 | 0 | 0 | 0 | 0 | … |
| A4GALT | 0 | 0 | 0 | 0 | 0 | … |
| AAAS | 0 | 37 | 30 | 21 | 0 | … |
| AACS | 0 | 0 | 0 | 0 | 2 | … |
| AADAT | 0 | 0 | 0 | 0 | 0 | … |
| … | … | … | … | … | … | … |
第二个是样本注释信息文件,命名为”cellAnnotations.txt”。一共两列,第一列是对应第一个文件的列名,第二列是细胞的分组
1 | MGH54_P2_C12 Microglia/Macrophage |
第三个是基因位置信息文件,命名为”geneOrderingFile.txt”。一共四列,第一列对应第一个文件的行名,其余三列则是基因的位置。注:基因名不能有重复
1 | WASH7P chr1 14363 29806 |
两步法
最复杂的工作就是准备输入文件,而一旦上述三个文件已经创建完成,那么分析只要两步以及根据结果对参数进行调整。
第一步,根据上述的三个文件创建inferCNV对象
1 | infercnv_obj = CreateInfercnvObject(raw_counts_matrix=matrix, # 可以直接提供矩阵对象 |
这一步的一个关键参数是ref_group_name, 用于设置参考组。假如你并不知道哪个组是正常,哪个组不正常,那么设置为ref_group_name=NULL, 那么inferCNV会以全局平均值作为基线,这适用于有足够细胞存在差异的情况。此外,这里的raw_count_matrix是排除了低质量细胞的count矩阵。
第二步,运行标准的inferCNV流程。
1 | # perform infercnv operations to reveal cnv signal |
关键参数是cutoff, 用于选择哪些基因会被用于分析(在所有细胞的平均表达量需要大于某个阈值)。这个需要根据具体的测序深度来算,官方教程建议10X设置为0.1,smart-seq设置为1。你可以先评估下不同阈值下的保留基因数,决定具体值。cluster_by_groups用于声明是否根据细胞注释文件的分组对肿瘤细胞进行分群。
最终会输出很多文件在out_dir的目录下,而实际有用的是下面几个
- infercnv.preliminary.png : 初步的inferCNV展示结果(未经去噪或HMM预测)
- infercnv.png : 最终inferCNV产生的去噪后的热图.
- infercnv.references.txt : 正常细胞矩阵.
- infercnv.observations.txt : 肿瘤细胞矩阵.
- infercnv.observation_groupings.txt : 肿瘤细胞聚类后的分组关系.
- infercnv.observations_dendrogram.txt : NEWICK格式,展示细胞间的层次关系.
参数说明
Infercnv::run的参数非常之多,总体上分为如下几类
- 基本设置
- 平滑参数
- 基于肿瘤亚群的下游分析(HMM或non-DE-masking)
- 根据 BayesNet P(Normal) 过滤低可信度HMM预测结果
- 肿瘤亚群细分
- 去噪参数
- 离群值修剪
- 其他选项
- 实验性参数(不稳定)
- 差异表达分析的实验性参数
你可以按照具体的需求修改不同步骤的参数,例如聚类默认cluster_by_groups=FALSE会根据k_obs_groups聚类成指定的组数,而层次聚类方法用于计算组间相似度的参数则是hclust_method.
此外,设置HMM=TRUE 的计算时间会长于HMM=FALSE,因此可以先设置HMM=FALSE快速查看结果。
在运行过程中它会显示每个步骤的信息,官方文档给出了示意图帮助理解。
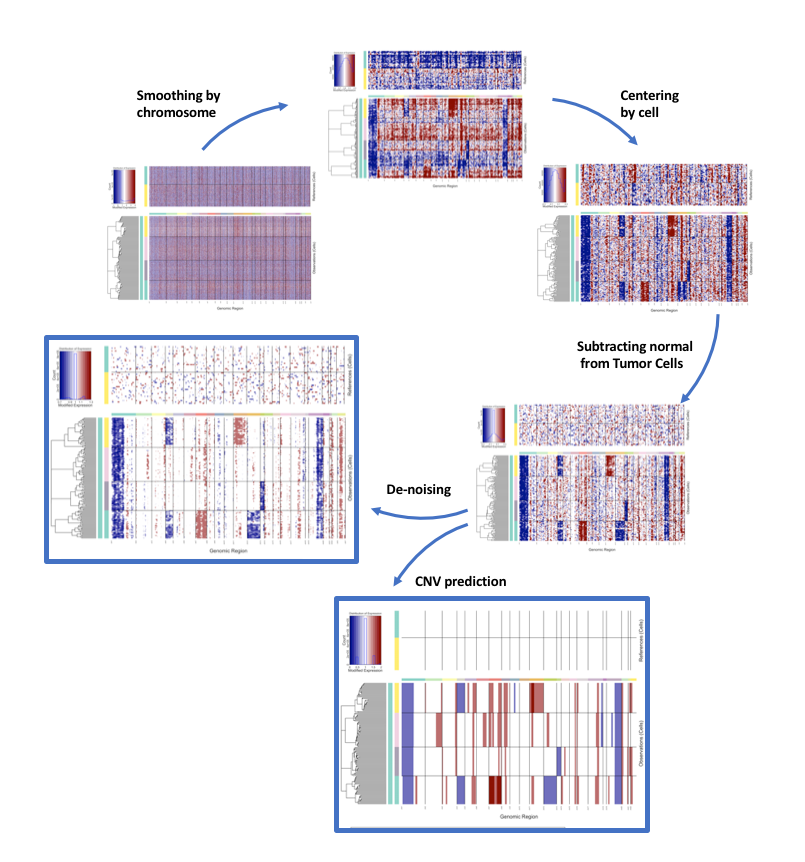
提取信息
inferCNV会输出一个” map_metadata_from_infercnv .txt”文件用于记录每个细胞的元信息,所有信息都可以从该文件中进行提取。或者使用infercnv::add_to_seurat将信息直接增加到原来的seurat对象中。
参考资料
- 软件安装: https://github.com/broadinstitute/inferCNV/wiki/Installing-infercnv
- 文件定义: https://github.com/broadinstitute/inferCNV/wiki/File-Definitions
- 运行inferCNV: https://github.com/broadinstitute/inferCNV/wiki/Running-InferCNV
- 引用: inferCNV of the Trinity CTAT Project. https://github.com/broadinstitute/inferCNV
关于inferCNV的算法原理在如下几篇文章中有说明
- Anoop P. Patel, Itay Tirosh, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014 Jun 20: 1396-1401
- Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016 Apr 8;352(6282):189-96
- Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016 Nov 10;539(7628):309-313. PubMed PMID: 27806376; PubMed Central PMCID: PMC5465819.
- Venteicher AS, Tirosh I, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017 Mar 31;355(6332).PubMed PMID: 28360267; PubMed Central PMCID: PMC5519096.
- Puram SV, Tirosh I, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017 Dec 14;171(7):1611-1624.e24. PubMed PMID: 29198524; PubMed Central PMCID: PMC5878932.