SCALE全称是Single-Cell ATAC-seq analysis vie Latent feature Extraction, 从名字中就能知道这个软件是通过隐特征提取的方式分析单细胞ATAC-seq数据。
在文章中,作者从开发者的角度列出了目前的scATAC-seq分析软件,chromVAR, scABC, cisTopic, scVI,发现每个软件都有一定的不足之处,而从我们软件使用者的角度,其实可以考虑都试试这些工具。
SCALE结合了深度生成模型(Depp Generative Models)变分自动编码器框架(Variational Autoencoder, VAE)与概率高斯混合模型(Gaussian Mixture Model, GMM)去学习隐特征,用于准确地鉴定scATAC-seq数据中的特征。
文章通过一张图来解释了软件的工作机制:
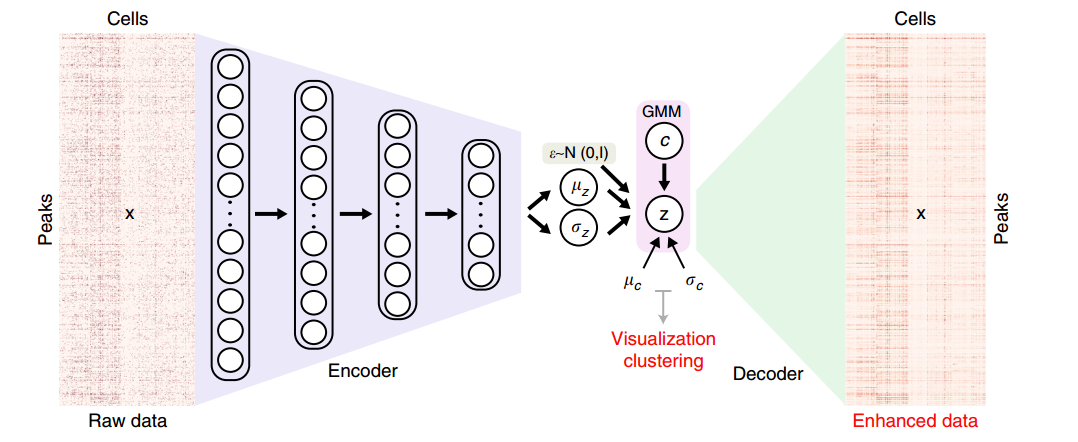
SCALE将sc-ATAC-seq的输入数据x(Cells-by-Peaks矩阵)建模成一个联合分布,p(x,z,c),c是GMM组件中对应的预定义的K个聚类,z是一个隐变量,是细胞在所有peak中实际可能的值,用于后续的聚类和可视化。z通过$z = \mu_z + \sigma_Z \times \epsilon$ 计算而得,公式里面的 $\mu_z$ $\sigma_z$ 是编码器网络从x中学习而得,$\epsilon$ 则是从 $\mathbb{N}(0,\mathbf{I})$ 抽样而成。
从公式中我们还可以发现z其实和GMM的c有关,所以p(x,z,c)也可以写成P(x|z)p(z|c)p(c),而p(c)是K个预定义聚类分布的离散概率分布,p(z|c)服从混合高斯分布,而p(x|z)则是服从多变量伯努利分布(multivartiable Bernoulli distribution), 通过解码者网络建模而成。
当然从一个软件使用者的角度而言,我们不会去关心代码,也不会关心原理,我们更关心的是这个工具能做什么。SCALE能做以下的分析
- SCALE可以对隐特征聚类识别细胞类群
- SCALE可以降噪,恢复缺失的peak
- SCALE能够区分批次效应和生物学细胞类群之间的差异
软件安装
推荐使用conda的方式进行软件安装(我测试过了,运行没有问题)
第一步:创建一个环境,名字就是SCALE,并且启动该环境
1 | conda create -n SCALE python=3.6 pytorch |
第二步:从GitHub上克隆该项目
1 | git clone git://github.com/jsxlei/SCALE.git |
第三步:安装SCALE
1 | cd SCALE |
之后分析的时候,只需要通过conda activate SCALE就能启动分析环境。
考虑后续要交互的读取数据和可视化,那么建议再安装一个Jupyter
1 | conda install jupyter |
软件使用
SCALE支持两类输入文件:
- count矩阵,行为peak,列为barcode
- 10X输出文件: count.mtx.gz, peak.tsv, barcode.tsv
我们以官方提供的Forebrain数据集为例进行介绍,因为这个数据相对于另外一个数据集Mouse Atlas小多了。
我们在服务器上新建一个文件夹,用于存放从https://cloud.tsinghua.edu.cn/d/bda0332212154163a647/下载的数据
1 | mkdir Forebrain |
保证Forebrain有下载好的数据
1 | $ ls Forebrain |
之后运行程序
1 | SCALE.py -d Forebrain/data.txt -k 8 --impute |
软件运行步骤为:
- 加载数据: Loading data
- 模型训练: Training Model
- 输出结果: Saving imputed data
其中模型训练这一步时间比较久,可以尝试用GPU加速(我是普通CPU服务器没有办法)。最终会在当前文件夹看到一个output文件夹,里面有如下内容:
- imputed_data.txt: 每个细胞在每个特征的推断值,建议用
--binary保存二进制格式 - model.pt: 用于重复结果的模型文件,
--pretrain参数能够读取该模型 - feature.txt: 每个细胞的隐特征,用于聚类和可视化
- cluster_assignments.txt: 两列,barcode和所属类群
- tsne.txt, tsne.pdf: tSNE的坐标和PDF文件,坐标文件可以导入到R语言进行可视化
上面是命令行部分,下面则是Python环境进行交互式操作,输入jupyter notebook,之后在网页上打开
首先是导入各种Python库
1 | import pandas as pd |
然后加载分析结果,包括聚类信息和特征信息
1 | y = pd.read_csv('output/cluster_assignments.txt', sep='\t', index_col=0, header=None)[1].values |
通过热图展示不同聚类细胞之间的差异图
1 | plot_heatmap(feature.T, y, |
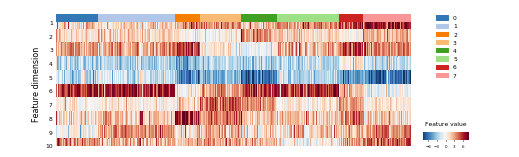
如果要矫正批次效应,可以通过根据feature的heatmap,去掉和batch相关的feature来实现
我们可以展示SCALE对原始数据纠正后的值(imputed data), 该结果也能提高chromVAR鉴定motif的效果
1 | imputed = pd.read_csv('output/imputed_data.txt', sep='\t', index_col=0) |
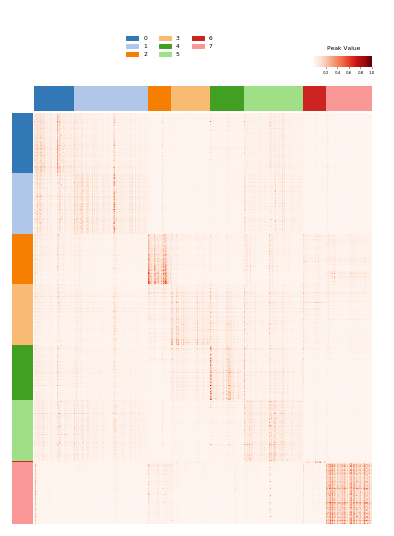
展示聚类特异性的peak, 分析由mat_specificity_score和cluster_specific完成
1 | from scale.specifity import cluster_specific, mat_specificity_score |
参数介绍
通过SCALE.py -h可以输出SCALE的所有可用参数
-d/--dataset: 单个文件矩阵应该指定文件路径,10X输出的多个文件则是文件目录-k: 设定输出结果的聚类数-o: 输出文件路径--pretrain: 读取之前训练的模型--lr: 修改起始学习速率, 默认是0.002,和模型训练有关--batch_size: 批处理大小, 默认就行,不需要修改(和批次效应处理无关)-g GPU: 选择GPU设备数目,非GPU服务器用不到--seed: 初始随机数种子,通常在遇到nan缺失时考虑修改-encode_dim,-decode_dim: 编码器和解码器的维度,通常也不需要修改-latent隐藏层维度--low,--high: 过滤低质量的peak, 即出现比例高于或者低于某个阈值的peak,默认是0.01和0.9。作者推荐保留1万-3万的peak用于SCALE分析。如果数据质量很高,且peak数不多于10万,那么可以不过滤。--min_peaks: 过滤低质量细胞,如果该细胞的peak低于阈值log_transform:log2(x+1)的变换--max_iter: 最大迭代数,默认是30000, 可以观察损失收敛的情况来修改,也就是训练模型这一步输出的信息-weight_decay: 没有说明--impute: 保存推断数据,默认开启--binary: 推荐加上该参数,减少imputed data占用空间--no_tsne: 不需要保存t-SNE结果--reference: 参考细胞类型-t: 如果输出矩阵是列为peak,行为barcode,用该参数进行转置
对于使用者而言,我们一般只用修改-k更改最后的聚类数,--low, --high, ---min_peaks来对原始数据进行过滤,以及加上--binary节约空间。
假如在训练模型阶段,发现输出信息为loss=nan recon_loss=nan kl_loss=nan,十有八九最终会报错退出, 可以如下的参数调整
- 更改
--seed - 用更加严格的条件过滤peak,例如
-x 0.04或-x 0.06 - 降低初始的学习速率,
--lr 0.0002