基因组组装完成后,或者是完成了草图,就不可避免遇到一个问题,需要对基因组序列进行注释。注释之前首先得构建基因模型,有三种策略:
- 从头注释(de novo prediction):通过已有的概率模型来预测基因结构,在预测剪切位点和UTR区准确性较低
- 同源预测(homology-based prediction):有一些基因蛋白在相近物种间的保守型搞,所以可以使用已有的高质量近缘物种注释信息通过序列联配的方式确定外显子边界和剪切位点
- 基于转录组预测(transcriptome-based prediction):通过物种的RNA-seq数据辅助注释,能够较为准确的确定剪切位点和外显子区域。
每一种方法都有自己的优缺点,所以最后需要用EvidenceModeler(EVM)和GLEAN工具进行整合,合并成完整的基因结构。基于可靠的基因结构,后续可才是功能注释,蛋白功能域注释,基因本体论注释,通路注释等。
那么基因注释重要吗?可以说是非常重要了,尤其是高通量测序非常便宜的现在。你可以花不到一万的价格对600M的物种进行100X的普通文库测序,然后拼接出草图。但是这个草图的价值还需要你进行注释后才能显现出来。有可能你和诺贝尔奖就差一个注释的基因组。
从案例中学习套路
陆地棉基因组注释
文章标题为“Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement”.
同源注释:从Phytozome上下载了7个植物的基因组蛋白序列(Arabidopsis thaliana, Carica papaya, Glycine max, G. raimondii, Populus trichocarpa, Theobroma cacao and Vitis vinifera), 使用 TblastN 将蛋白序列比对到组装序列上,E-value的阈值为1e-5. 将不同蛋白的BLAST的hits用 Solar 软件进行合并。GeneWise 根据每个BLAST hit的对应基因区域预测完整的基因结构。
从头预测:先得构建repeat-mask genome, 在这个基础上就用 August, Genescan, GlimmerHMM, Geneid 和 SNAP 预测编码区
转录组预测:用Tophat将RNA-seq数据比对到组装序列上,然后用cufflinks组装转录本形成基因模型。
综上,使用 EvidenceModeler(EVM) 将上面的结果组装成非冗余的基因结构。进一步根据Cscore > 0.5,peptide coverage > 0.5 和CDS overlaping with TE进行筛选。还有过滤掉超过30%编码区被Pfam或Interprot TE domain的注释的基因模型。
这些基因模型使用BLASTP进行功能注释,所用数据库为SWiss-Prot和TrEMBL.蛋白功能使用InterProScan和HMMER注释,数据库为InterPro和Pfam。GO注释则是直接雇佣InterPro和Pfam注释得到的对应entry。通路注释使用KEGG数据库。
Cardamine hirsuta基因组注释
文章标题为“The Cardamine hirsuta genome offers insight into the evolution of morphological diversity”。
同源注释:使用 GenomeThreader 以拟南芥为剪切模型,以及PlantsGDB resourc上 Brassica rapa (v1.1), A. thaliana(TAIR10), A. lyrata (v6), tomato (v3.6), poplar (v2) 和 A. thaliana (version PUT-169), B. napus (version PUT-172) EST assemblies 的完整的代表性蛋白集。
转录本预测: 将 C. hirsuta RNA-seq数据比对到基因序列,然后用cufflinks拼接
从头预测:转录本预测得到的潜在蛋白编码转录本使用网页工具 ORFpredictor 进行预测, 同时用 blastx 和 A. thalina 进行比较,选择90%序列相似度和最高5%长度差异的部分从而保证保留完整的编码框(有启动子和终止子)。 这些基因模型根据相互之间的相似度和重叠度进行聚类,高度相似(>95)从聚类中剔除,保证非冗余训练集。为了训练gene finder, 它们选随机选取了2000个位点,20%是单个外显子基因。从头预测工具为 August , GlimmerHMM, Geneid 和 SNAP . 此外还用了Fgenesh+, 以双子叶特异矩阵为参数进行预测。
最后使用JIGSAW算法根据以上结果进行训练,随后再次用JIGSAW对每个基因模型计算统计学权重。
可变剪切模型则是基于苗、叶、花和果实的RNA-seq比对组装结果。
GO注释使用AHRD流程
小结
举的2个例子都是植物,主要是植物基因组不仅是组装,注释都是一大难题。因为植物基因组有大量的重复区,假基因,还有很多新的蛋白编码基因和非编码基因,比如说玉米基因组80%以上都是重复区域。然后当我检索这两篇文章所用工具的时候,我不经意或者说不可避免就遇到了这个网站 http://www.plantgdb.org/ , 一个整合植物基因组学工具和资源的网站,但是这个网站似乎2年没有更新了。当然这个网站也挺不错,http://bioservices.usd.edu/gsap.html, 他给出了一套完整的注释流程以及每一步的输入和输出情况。
此外,2017年在《Briefings in Bioinformatics》发表的”Plant genome and transcriptome annotations: from misconceptions to simple solution” 则是从五个角度对植物基因组注释做了很完整的总结
- 植物科学的常见本体
- 功能注释的常用数据库和资源
- 已注释的植物基因组意味着什么
- 一个自动化注释流程
- 一个参考流程图,用来说明使用公用数据库注释植物基因组/转录组的常规步骤
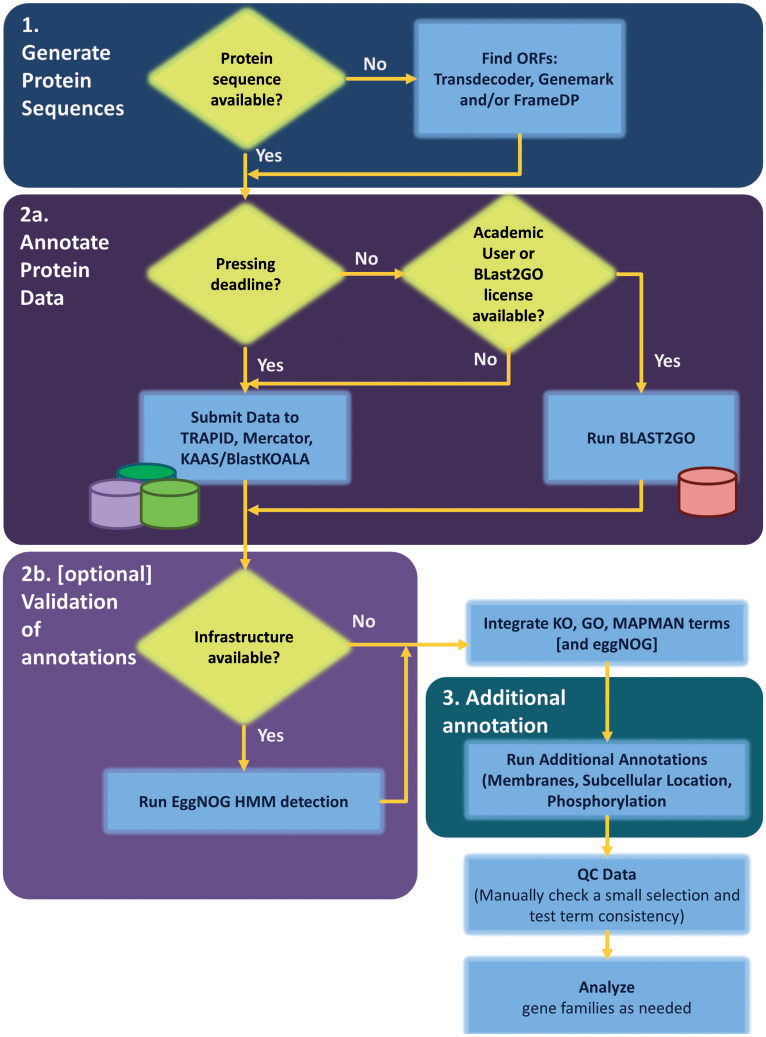
以上,通过套路我们对整个基因组注释有一个大概的了解,后续就需要通过实际操作来理解细节。
基因组注释
当我们谈到基因注释的时候,我们通常认为注释是指“对基因功能的描述”,比如说A基因在细胞的那个部分,通过招募B来调控C,从而引起病变。但是基因结构也是注释的一种形式,而且是先决条件,也就是在看似随机的ATCG的碱基排列中找到特殊的部分,而这些特殊的区域有着不一样的功能。
在正式启动基因组注释项目之前,需要先检查组装是否合格,比如contig N50的长度是否大于基因的平均长度,使用BUSCO/CEGMA检查基因的完整性,如果不满足要求,可能输出结果中大部分的contig中都不存在一个完整的基因结构。当组装得到的contig符合要求时,就可以开始基因组注释环节,这一步分为三步:基因结构预测,基因功能注释,可视化和质控。
基因组结构注释
基因结构注释应是功能注释的先决条件,完整的真核生物基因组注释流程需要如下步骤:
- 必要的基因组重复序列屏蔽
- 从头寻找基因, 可用工具为: GeneMarkHMM, FGENESH, Augustus, SNAP, GlimmerHMM, Genscan
- 同源蛋白预测, 内含子分析: GeneWIse, Exonerate, GenomeThreader
- 将EST序列,全长cDNA序列和Trinity/Cufflinks/Stringtie组装的转录组和基因组联配
- 如果第4步用到了多个数据来源,使用PASA基于重叠情况进行联配
- 使用EvidenceModler根据上述结果进行整合
- 使用PASA更新EVM的一致性预测,增加UTR注释和可变剪切注释
- 必要的人工检查
基本上是套路化的分析流程,也就有一些工具通过整合几步开发了流程管理工具,比如说BRAKER结合了GeneMark和Augustus,MAKER2整合了SNAP,Exonerate,虽然BRAKER说自己的效果比MAKER2好,但是用的人似乎不多,根据web of knowledge统计,两者的引用率分别是44,283, 当然BRAKER是2016,MAKER2是2011,后者在时间上有优势。
这里准备先按部就班的按照流程进行注释,所用的数据是 Cardamine hirsuta , 数据下载方式如下
1 | # Cardamine hirsutat基因组数据 |
软件安装不在正文中出现,会放在附录中,除了某些特别复杂的软件。
01-重复序列屏蔽
重复屏蔽:真核生物的基因组存在大量的重复序列,植物基因组的重复序列甚至可以高达80%。尽管重复序列对维持染色体的空间结构、基因的表达调控、遗传重组等都具有重要作用,但是却会导致BLAST的结果出现大量假阳性,增加基因结构的预测的计算压力甚至影响注释正确性。基因组中的重复按照序列特征可以分为两类:串联重复(tandem repeats)和散在重复(interspersed repeats).
鉴定基因组重复区域的方法有两种:一种基于文库(library)的同源(homology)方法,该文库收集了其他物种的某一种重复的一致性序列,通过相似性来鉴定重复;另一种是从头预测(de novo),将序列和自己比较或者是高频K-mer来鉴定重复。
目前重复序列注释主要软件就是RepeatMasker和RepeatModel。这里要注意分析的fasta的ID不能过长,不然会报错。如果序列ID过长可以使用bioawk进行转换,后续用到RepatModel不支持多行存放序列的fasta格式。
直接使用同源注释工具RepeatMasker寻找重复序列:
1 | mkdir 00-RepeatMask |
输出结果中主要关注如下三个(其中xxx表示一类文件名)
- xxx.fa.masked, 将重复序列用N代替
- xxx.fa.out.gff, 以gff2形式存放重复序列出现的位置
- xxx.fa.tbl, 该文件记录着分类信息
1 | cat 00-RepeatMask/chi_unmasked.fa.tbl |
也就是说该物种198M中有将近18%的重复序列,作为参考,拟南芥125Mb 14%重复序列, 水稻389M,36%重复,人类基因组是3G,50%左右的重复序列。
使用最后的chi_unmasked.fa.masked用于下一步的基因结构预测。
注:当然也可以用RepeatModel进行从头预测,得到的预测结果后续可以整合到RepeatMasker
1 | # de novo predict |
这一步速度极其慢,由于我们的目的只是获取屏蔽后序列降低后续从头预测的压力,所以可以先不做这一步。在后续分析重复序列在基因组进化上的作用时可以做这一步。下
如果从头预测的结果与同源预测的结果有30%以上的overlap,并且分类不一致,会把从头预测的结果过滤掉。从头预测与同源预测结果有overlap,但是分类一致的,都会保留。但是统计的时候不会重复统计。
02-从头(ab initio)预测基因
基于已有模型或无监督训练
目前的从头预测软件大多是基于HMM(隐马尔科夫链)和贝叶斯理论,通过已有物种的注释信息对软件进行训练,从训练结果中去推断一段基因序列中可能的结构,在这方面做的最好的工具是AUGUSTUS 它可以仅使用序列信息进行预测,也可以整合EST, cDNA, RNA-seq数据作为先验模型进行预测。
AUGUSTUS的无root安装比较麻烦,我折腾了好几天最后卒,不过辛亏有bioconda,conda create -n annotation augustus=3.3.
它的使用看起来很简单,我们可以尝试使用一段拟南芥已知的基因序列让其预测,比如前8k序列
1 | seqkit faidx TAIR10.fa Chr1:1-8000 > test.fa |
如果仅仅看两者的CDS区,结果完全一致,相当于看过一遍参考答案去做题目,题目都做对了。
注:已经被训练的物种信息可以用
augustus --species=help查看。
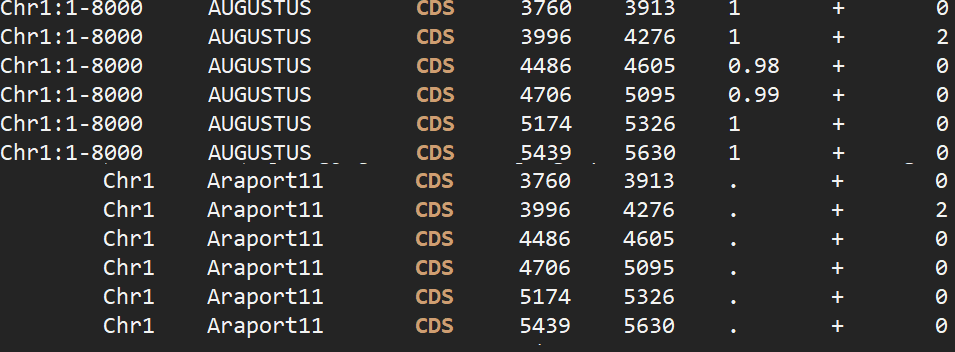
在不使用RNA-seq数据的情况下,可以基于拟南芥的训练模型进行预测,采用下面的方式多条染色体并行augustus
1 | mkdir 01-augustsus && cd 01-augustsus |
AUGUSTUS依赖于已有的模型,而GeneMark-ES/ET则是唯一一款支持无监督训练模型,之后再识别真核基因组蛋白编码区的工具。
1 | gmes_petap.pl --ES --sequence genome.fa --cores 50 |
最后得到的是genemark.gtf,是标准的GTF格式,可以使用Sequence Ontology Project提供的gtf2gff3.pl进行转换
1 | wget http://genes.mit.edu/burgelab/miso/scripts/gtf2gff3.pl |
不同从头预测软件的实际效果可以通过在IGV中加载文章提供的gff文件和预测后的gff文件进行比较,一般会存在如下几个问题:
- 基因多了,或者少了,也就是假阳性和假阴性现象
- UTR区域难以预测,这个比较正常
- 未正确识别可变剪切位点,导致前后几个基因识别成一个基因
考虑到转录组测序已经非常便宜,可以通过该物种的RNA-seq提供覆盖度信息进行预测。
基于转录组数据预测
根据已有的模型或者自训练可以正确预测很大一部分的基因,但如果需要提高预测的正确性,还需要额外的信息。在过去就需要提供物种本身的cDNA, EST,而现在更多的是基于转录组序列进行训练。尽管RNA-seq数据在基因组上的比对情况能够推测出内含子位置,根据覆盖度可以推测出外显子和非编码区的边界,但是仅仅依赖于RNA-seq的覆盖不能可信地推测出蛋白编码区(Hoff K.J. Stanke M. 2015).
AUGUSTUS可以利用转录组比对数据中的位置信息来训练模型,GeneMark-ET可以利用RNA-seq得到的内含子位点信息自我训练HMM参数,进行基因预测。BRAKER2将两者进行整合,使用GeneMark-ET根据RNA-seq无监督训练模型寻找基因,然后用AUGUSTUS进行模型训练,最后完成基因预测
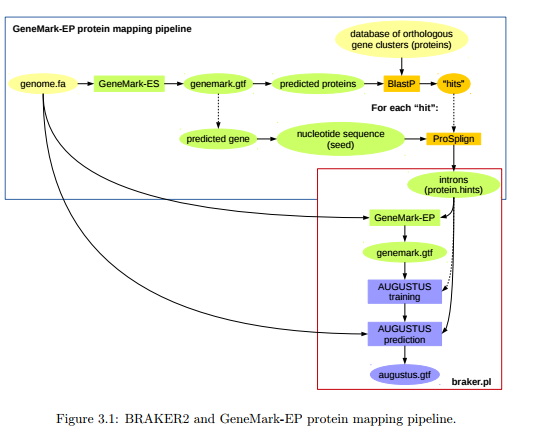
首先使用hisat2根据屏蔽后的参考序列建立索引,进行比对。
1 | # 项目根目录 |
然后,以未屏蔽重复序列的参考序列和BAM文件作为输入,让BRAKER2(安装会稍显麻烦,因为依赖许多软件)进行预测。
1 | braker.pl --gff3 --cores 50 --species=carhr --genome=chi_unmasked.fa --bam=02-barker/leaf_ox_r1.bam,02-barker/ox_flower16.bam,02-barker/ox_flower9.bam |
最后会得到如下输出文件
- hintsfile.gff: 从RNA-seq比对结果的BAM文件中提取,其中内含子用于训练GeneMark-EX, 使用所有特征训练AUGUSTUS
- GeneMark-ET/genemark.gtf: GeneMark-EX根据RNA-seq数据训练后预测的基因
- augustus.hints.gff: AUGUSTUS输出文件
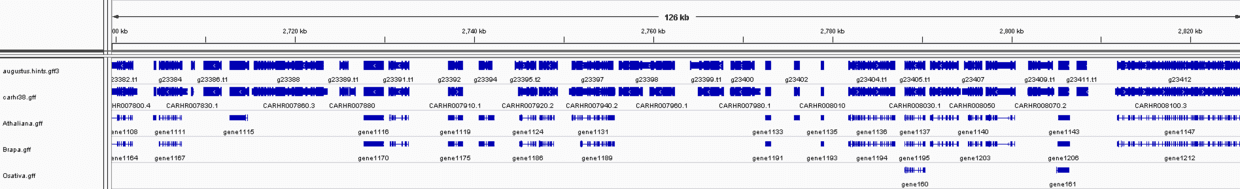
将augustus.hints.gff3和文章的注释文件(carhr38.gtf)比较,见下图:

其实不难发现,在不考虑UTR区域情况下,两者的差别其实更多表现是基因数目上,其实也就是利用转录组数据推测结构的问题所在,没有覆盖的区域到底是真的没有基因,还是有基因结构只不过所用组织没有表达,或者说那个区域其实是假基因?此外,如果基因间隔区域很短,有时候还会错误地把两个不同的基因预测为一个基因。因此,应该注重RNA-seq数据在剪切位点识别和外显子边界确定的优势。
03-同源预测基因结构
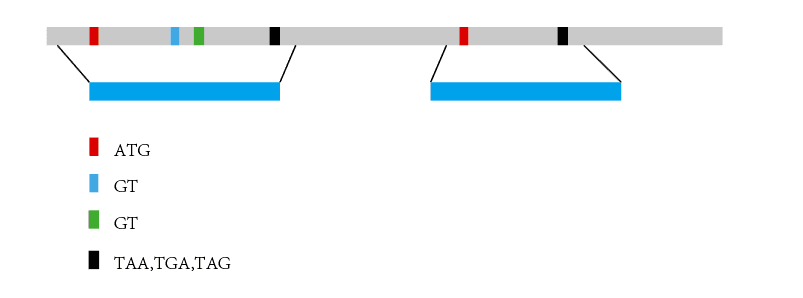
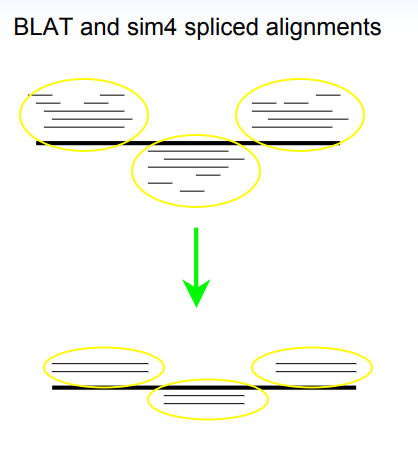
同源预测(homology prediction)利用近缘物种已知基因进行序列比对,找到同源序列。然后在同源序列的基础上,根据基因信号如剪切信号、基因起始和终止密码子对基因结构进行预测,如下示意图:

相对于从头预测的“大海捞针”,同源预测相当于先用一块磁铁在基因组大海中缩小了可能区域,然后从可能区域中鉴定基因结构。在10年之前,当时RNA-seq还没有普及, 只有少部分物种才有EST序列和cDNA序列的情况下,这的确是一个比较好的策略,那么问题来了,现在还需要进行这一步吗,如果需要是出于那种角度考虑呢?
在同源预测上,目前看到的大部分基因组文章都是基于TBLASTN + GeneWise,这可能是因为大部分基因组文章都是国内做的,这些注释自然而言用的就是公司的流程,然后目前国内的公司大多数又和某一家公司有一些关系。不过最近的3010水稻泛基因组用的是MAKER, 感谢部分提到这部分工作是由M. Roa(Philippine Genome Center Core Facilities for Bioinformatics, Department of Science)做的,算是一股清流吧。当然我在看Cardamine hirsuta基因组注释文章,发现它们同源注释部分用的是GenomeThreader, 该工具在本篇文章成文时的3月之前又更新了。
GeneWise的网站说它目前由Ewan Birney维护,只不过不继续开发了,因为Guy Slater开发Exonerate解决了GeneWise存在的很多问题,并且速度快了1000倍。考虑到目前只有GeneWise能利用HMM根据蛋白找DNA,而且ENSEMBL的注释流程也有一些核心模块用到了它,所以作者依旧在缓慢的开发这个工具(自2.4.1已经10多年没有更新了),当然这个工具也是非常的慢。尽管这一步不会用到GeneWise作为我们的同源注释选项,但是我们可以尝试用GeneWise手工注释一个基因,主要步骤如下
- 第一步: 使用BLASTX,根据dna序列搜索到蛋白序列,只需要第一个最佳比对结果
- 第二步: 选择最佳比对的氨基酸序列
- 第三步: 将dna序列前后延长2kb,与氨基酸序列一并传入给genewise进行同源预测
提取前5K序列,然后选择在TAIR上用BLASTX进行比对
1 | seqkit faidx chi_unmasked.fa Chr1:1-5000 > chr1_5k.fa |
选择第一个比对结果中的氨基酸序列,和前5k的DNA序列一并作为GeneWise的输入
最后的结果出乎了我的意料
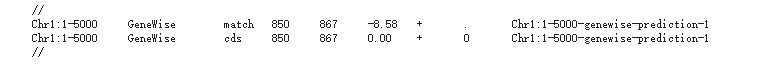
让我们跳过这个尴尬的环节,毕竟很可能是我不太熟练使用工作所致。这里说点我的看法,除非你真的没有转录组数据,必须要用到同源物种的蛋白进行预测,或者你手动处理几个基因,否则不建议使用这个工具,因为你可能连安装都搞不定。
让我们用GenomeThreader基于上面的DNA序列和氨基酸序列进行同源基因结构预测吧
1 | gth -genomic chr1_5k.fa -protein cer.fa -intermediate -gff3out |
结果一致,并且从RNA-seq的覆盖情况也符合预期
1 | Chr1 gth exon 1027 1197 Parent=gene1 Chr1 MIPS_CARH_v3.8 exon 1027 1197 |
全基因组范围预测流程如下:
准备cDNA和或protein序列:在https://phytozome.jgi.doe.gov/p下载靠谱的物种的蛋白质序列,如 Arabidopsis thaliana, Oryza sativa, Brassica rapa, 查找文献寻找目前该物种的已有EST/cDNA序列,或者RNA-seq从头组装转录组。这里仅考虑用同源物种的蛋白序列进行比对分析,转录组从头组装数据用于PASA整体比对到参考基因组和更新已有的基因解雇。
分别测试下不同物种的同源注释结果
1 | #run seperately |
在定性角度上来看,同源注释的结果和从头预测的没啥差别, 其中B. rapa和A. thaliana和C. hirsuta都属于十字花科,而O. sativa是禾本科, 所以前两者预测的效果好。
当然实际的同源注释流程中不能是单个物种分别预测,应该是将所有的蛋白序列进行合并,然后用BLASTX找到最优的联配,之后用GenomeThreader进行预测。PASA流程提到的UniRef90作为同源注释的搜索数据库可能是更好的选择,由于UniRef优先选择哪些人工审查、注释质量高、来源于模式动植物的蛋白,所以可靠性相对于直接使用同源物中可能更高。
BLASTX + GenomeThreader的代码探索中
04-RNA-seq的两种使用策略
对于RNA-seq数据,有两种使用策略,一种是使用HISAT2 + StringTie先比对再组装, 一种是从头组装,然后使用PASA将转录本比对到基因组上。
基于HISAT2 + StringTie
首先,使用HISAT2将RNA-seq数据比对到参考基因组, 这一步和之前相似,但是要增加一个参数--dta,使得StingTie能更好的利用双端信息
1 | hisat2-build 01-augustus/genome.fa index/chi_masked |
然后用StringTie进行转录本预测
1 | stringtie -p 10 -o rna-seq/merged.gtf rna-seq/merged.bam |
对于后续的EvidenceModeler而言,它不需要UTR信息,只需要编码区CDS,需要用TransDecoder进行编码区预测
1 | util/cufflinks_gtf_genome_to_cdna_fasta.pl merged.gtf input/chi_masked.fa > transcripts.fasta |
最后结果transcripts.fasta.transdecoder.gff3用于提供给EvidenceModeler
基于PASA
在多年以前,那个基因组组装还没有白菜价,只有几个模式物种基因组的时代,对于一个未测序的基因组,研究者如果要研究某一个基因的功能,大多会通过同源物种相似基因设计PCR引物,然后去扩增cDNA. 如果是一个已知基因组的物种,如果要大规模识别基因, 研究者通常会使用EST(expressed sequence tags)序列。
相对于基于算法的从头预测,cDNA和EST序列更能够真实的反应出一个基因的真实结构,如可变剪切、UTR和Poly-A位点。PASA(Progam to Assemble Spliced Alignments)流程最早用于拟南芥基因组注释,最初的设计是通过将全长(full-length)cDNA和EST比对到参考基因组上,去发现和更新基因组注释。其中FL-cDNA和EST序列对最后结果的权重不同。
这是以前的故事,现在的故事是二代转录组以及一些三代转录组数据,那么如何处理这些数据呢?我认为三代转录组相对于过去的FL-cDNA,而二代转录组数据经过拼接后可以看作是更长的EST序列。由于目前最普及的还是普通的mRNA-seq, 也就只介绍这部分流程。
考虑到我还没有研究过三代的全长转录组,分析过数据,这里的思考极有可能出错,后续可能会修改这一部分思考。
转录组组装使用Trinity(conda安装)
1 | cd rna-seq |
PASA是由30多个命令组成的流程,相关命令位于PASApipeline/scripts,为了适应不同的分析,有些参数需要通过修改配置文件更改,
1 | cp ~/opt/biosoft/PASApipeline/pasa_conf/pasa.alignAssembly.Template.txt alignAssembly.config |
上述几行配置文件表明SQLite3数据库的名字,设置了scripts/validate_alignments_in_db.dbi的几个参数, 表示联配程度和相似程度。后续以Trinity组装结果和参考基因组作为输入,运行程序:
1 | ~/opt/biosoft/PASApipeline/scripts/Launch_PASA_pipeline.pl -c alignAssembly.config -C -R -g ../chi_unmasked.fa -t ../rna-seq/trinity_out_dir/Trinity.fasta --ALIGNERS blat,gmap |
最后结果如下:
- database.sqlite.pasa_assemblies_described.txt
- database.sqlite.pasa_assemblies.gff3
- database.sqlite.pasa_assemblies.gtf
- database.sqlite.pasa_assemblies.bed
其中gff3格式用于后续的分析。
目前的一些想法, 将从头组装的转录本比对到参考基因组上很大依赖组装结果,所以和EST序列和cDNA相比,质量上还有一点差距。
05-整合预测结果
从头预测,同源注释和转录组整合都会得到一个预测结果,相当于收集了大量证据,下一步就是通过这些证据定义出更加可靠的基因结构,这一步可以通过人工排查,也可以使用EVidenceModeler(EVM). EVM只接受三类输入文件:
gene_prediction.gff3: 标准的GFF3格式,必须要有gene, mRNA, exon, CDS这些特征,用EVidenceModeler-1.1.1/EvmUtils/gff3_gene_prediction_file_validator.pl验证protein_alignments.gff3: 标准的GFF3格式,第9列要有ID信和和target信息, 标明是比对结果transcript_alignments.gff3:标准的GFF3格式,第9列要有ID信和和target信息,标明是比对结果
EVM对gene_prediction.gff3有特殊的要求,就是GFF文件需要反映出一个基因的结构,gene->(mRNA -> (exon->cds(?))(+))(+), 表示一个基因可以有多个mRNA,即基因的可变剪接, 一个mRNA都可以由一个或者多个exon(外显子), 外显子可以是非翻译区(UTR),也可以是编码区(CDS). 而GlimmerHMM, SNAP等
这三类根据人为经验来确定其可信度,从直觉上就是用PASA根据mRNA得到的结果高于从头预测。
第一步:创建权重文件,第一列是来源类型(ABINITIO_PREDICTION, PROTEIN, TRANSCRIPT), 第二列对应着GFF3文件的第二列,第三列则是权重.我这里用了三个来源的数据。
1 | mkdir 05-EVM && cd 05-EVM |
我觉得根据基因组引导组装的ORF的可信度高于组装后比对,所以得分和PASA差不多一样高。从头预测权重一般都是1,但是BRAKER可信度稍微高一点,可以在2~5之间。
第二步:分割原始数据, 用于后续并行. 为了降低内存消耗,–segmentsSize设置的大小需要少于1Mb(这里是100k), –overlapSize的不能太小,如果数学好,可用设置成基因平均长度加上2个标准差,数学不好,就设置成10K吧
1 | cat transcripts.fasta.transdecoder.genome.gff3 ../braker/carhr/augustus.hints.gff3 > gene_predictions.gff3 |
第三步:创建并行运算命令并且执行
1 | ~/opt/biosoft/EVidenceModeler-1.1.1/EvmUtils/write_EVM_commands.pl --genome ../chi_unmasked.fa --weights `pwd`/weights.txt \ |
第四步:合并并行结果
1 | ~/opt/biosoft/EVidenceModeler-1.1.1/EvmUtils/recombine_EVM_partial_outputs.pl --partitions partitions_list.out --output_file_name evm.out |
第五步:结果转换成GFF3
1 | ~/opt/biosoft/EVidenceModeler-1.1.1/EvmUtils/convert_EVM_outputs_to_GFF3.pl --partitions partitions_list.out --output evm.out --genome ../chi_unmasked.fa |
当前权重设置下,EVM的结果更加严格,需要按照实际情况调整,增加其他证据。
06-可选步骤
注释过滤:对于初步预测得到的基因,还可以稍微优化一下,例如剔除编码少于50个AA的预测结果,将转座子单独放到一个文件中(软件有TransposonPSI)。
这里基于gffread先根据注释信息提取所有的CDS序列,过滤出长度不足50AA的序列,基于这些序列过滤原来的的注释
1 | gffread EVM.all.gff -g input/genome.fa -y tr_cds.fa |
使用PASA更新EVM结果:EVM结果不包括UTR区域和可变剪切的注释信息,可以使用PASA进行更新。然而这部分已经无法逃避MySQL, 服务器上并没有MySQL的权限,我需要学习Perl脚本进行修改。因此基因结构注释到此先放一放。
07-基因编号
对每个基因实现编号,形如ABCD000010的效果,方便后续分析。如下代码是基于EVM.all.gff,使用方法为python gffrename.py EVM_output.gff prefix > renamed.gff.
1 |
|
一些经验
如果有转录组数据,没必须要使用太多的从头预测工具,braker2 加 GlimmerHMM可能就够用了, 更多是使用PASA和StringTie利用好转录组数据进行注释。
基因功能注释
基因功能的注释依赖于上一步的基因结构预测,根据预测结果从基因组上提取翻译后的 蛋白序列 和主流的数据库进行比对,完成功能注释。常用数据库一共有以几种:
- Nr:NCBI官方非冗余蛋白数据库,包括PDB, Swiss-Prot, PIR, PRF; 如果要用DNA序列,就是nt库
- Pfam: 蛋白结构域注释的分类系统
- Swiss-Prot: 高质量的蛋白数据库,蛋白序列得到实验的验证
- KEGG: 代谢通路注释数据库.
- GO: 基因本体论注释数据库
除了以上几个比较通用的数据库外,其实还有很多小众数据库,应该根据课题研究和背景进行选择。注意,数据库本身并不能进行注释,你只是通过序列相似性进行搜索,而返回的结果你称之为注释。因此数据库和搜索工具要进行区分,所以你需要单独下载数据库和搜索工具,或者是同时下载包含数据库和搜索工具的安装包。
注意,后续分析中一定要保证你的蛋白序列中不能有代表氨基酸字符以外的字符,比如说有些软件会把最后一个终止密码子翻译成”.”或者”*“
BLASTP
这一部分用到的数据库都是用BLASTP进行检索,基本都是四步发:下载数据库,构建BLASTP索引,数据库检索,结果整理。其中结果整理需要根据BLASTP的输出格式调整。
Nr的NCBI收集的最全的蛋白序列数据库,但是无论是用NCBI的BLAST还是用速度比较快DIAMOND对nr进行搜索,其实都没有利用好物种本身的信息。因此在RefSeq上下载对应物种的蛋白序列, 用BLASTP进行注释即可。
1 | # download |
Swiss-Prot里收集了目前可信度最高的蛋白序列,一共有55w条记录,数据量比较小,
1 | # download |
关于结果整理,已经有很多人写了脚本,比如说我搜索BLAST XML CSV,就找到了https://github.com/Sunhh/NGS_data_processing/blob/master/annot_tools/blast_xml_parse.py, 所以就不过多介绍。
InterProScan
下面介绍的工具是InterProScan, 从它的9G的体量就可以感受它的强大之处,一次运行同时实现多个信息注释。
- InterPro注释
- Pfam数据库注释(可以通过hmmscan搜索pfam数据库完成)
- GO注释(可以基于NR和Pfam等数据库,然后BLAST2GO完成,)
- Reactome通路注释,不同于KEGG
命令如下
1 | ./interproscan-5.29-68.0/interproscan.sh -appl Pfam -f TSV -i sample.fa -cpu 50 -b sample -goterms -iprlookup -pa |
-appl告诉软件要执行哪些数据分析,勾选的越多,分析速度越慢,Pfam就行。
KEGG
KEGG数据库目前本地版收费,在线版收费,所以只能将蛋白序列在KEGG服务器上运行。因此你需要在http://www.genome.jp/tools/kaas/选择合适的工具进行后续的分析。我上传的50M大小蛋白序列,在KEGG服务器上只需要运行8个小时,也就是晚上提交任务,白天回来干活。
附录
基因组注释的常用软件:
- 重复区域
- RepeatMasker:识别基因组中的可能重复
- RepeatModeler: 识别新的重复序列
- LTR-FINDER: http://tlife.fudan.edu.cn/ltr_finder/
- 从头预测
- Augustus
- Fgenesh
- 同源预测
- GeneWise
- Exonerate
- Trinity
- GenomeThreader
- 注释合并
- GLEAN:已经落伍于时代了
- EvidenceModeler: 与时俱进
- 流程
- PASA:真核生物基因的转录本可变剪切自动化注释项目,需要提供物种的EST或RNA-seq数据
- MAKER
- BRAKER1: 使用GeneMark-ET和AUGUSTUS基于RNA-Seq注释基因结构
- EuGene
- 可视化
- IGV
- JBrowse/GBrowse
参考文献和推荐阅读:
- NCBI真核生物基因组注释流程https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/
- 真核基因组注释入门: “A beginner’s guide to eukaryotic genome annotation”
- 二代测序注释流程:Comparative Gene Finding: “Annotation Pipelines for Next-Generation Sequencing Projects”
- 基因组转录组注释策略: “Plant genome and transcriptome annotations: from misconceptions to simple solution”
- 重复序列综述: “Repetitive DNA and next-generation sequencing: computational challenges and solutions”
- MAKER2教程: http://weatherby.genetics.utah.edu/MAKER/wiki/index.php/MAKER_Tutorial_for_WGS_Assembly_and_Annotation_Winter_School_2018
- 《生物信息学》 樊龙江: 第1-5章: 基因预测与功能注释
- 《NGS生物信息分析》 陈连福: 真核生物基因组基因注释
- JGS流程: https://genome.jgi.doe.gov/programs/fungi/FungalGenomeAnnotationSOP.pdf
环境准备
数据下载
1 | # Cardamine hirsutat基因组数据 |
软件安装
RepeatMasker: 用于注释基因组的重复区,需要安装RMBlast, TRF,以及在http://www.girinst.org注册以下载Repbase
安装RepeatMasker
1 | cd ~/src |
在上面的基础上安装RepeatModel
1 | # RECON |
BLAST,BLAST有两个版本可供选择, WuBLAST或者NCBI-BLAST,我个人倾向于NCBI-BLAST,并且推荐使用编译后二进制版本,因为编译实在是太花时间了
1 | cd ~/src |
AUGUSTUS: 可以说是最好的预测软件,使用conda安装
1 | source activate annotation |
GeneMark-ES/ET则是唯一一款支持无监督训练模型, 软件下载需要登记
1 | cd ~/src |
GlimmerHMM:
1 | cd ~/src |
SNAP: 基因从头预测工具,在处理含有长内含子上的基因组上表现欠佳
1 | # 安装 |
Exnerate 2.2: 配对序列比对工具,提供二进制版本, 功能类似于GeneWise,能够将cDNA或蛋白以gao align的方式和基因组序列联配。
1 | cd ~/src |
GenomeThreader 1.70: 同源预测软件,1.7.0版本更新于2018年2月
1 | wget -4 http://genomethreader.org/distributions/gth-1.7.0-Linux_x86_64-64bit.tar.gz |
BRAKER2: 依赖AUGUSTUS 3.3, GeneMark-EX 4.33, BAMTOOLS 2.5.1, NCBI BLAST+ 2.2.31+(可选 SAMTOOLS 1.74+, GenomeThreader 1.70)
1 | cpan File::Spec::Functions Module::Load::Conditional POSIX Scalar::Util::Numeric YAML File::Which Logger::Simple Parallel::ForkManager |
TransDecoder 编码区域预测工具,需要预先安装NCBI-BLAST
1 | cpan URI::Escape |
MARKER: 使用conda安装会特别的方便,最好新建环境
1 | conda create -n marker marker |
PASA: 依赖于一个数据库(MySQL或SQLite), Perl模块(DBD::mysql或DBD::SQLite), GMAP, BLAT, Fasta3。由于MySQL在HPC集群中的表现不如SQLite,以及安装MySQL还需要各种管理员权限,于是就有人进行了修改,增加了feature/sqlite分支, 见Add support for SQLite
1 | cpan DB_File URI::Escape DBI DBD::SQLite |
EVidenceModeler: 整合不同来源的注释结果,找到可靠的基因结构
1 | cd ~/src |