重复序列注释有两种常用策略,基于同源序列相似性和基于重复序列结构特征。RepeatMasker是基于同源序列相似性注释序列的常用工具, RepeatModeler可用来从头对基因组的重复序列家族进行建模注释,它的核心组件是RECON和RepatScout。
这篇教程介绍如何使用RepeatModeler从头鉴定基因组的重复序列,之后用RepeatMasker根据自定义的重复序列库注释基因组的重复序列。
软件安装
原本的RepeatMasker和RepeatModeler的手动安装需要配置很多文件,但是利用bioconda就只用一行命令。
1 | conda create -n repeat repeatmasker repeatmodeler |
之后在RepeatMasker环境下配置运行环境。由于我的miniconda装在~/opt路径下,因此对应的RepeatMasker路径为~/opt/miniconda3/envs/repeat/share/RepeatMasker/
1 | conda activate repeat |
这一步只需要配置好比对软件
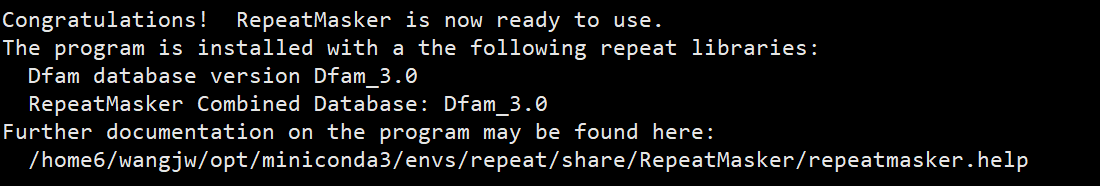
之后就会显示RepeatMasker已经配置完毕,其中Dfam_3.0是用于注释的数据库。
软件运行
以拟南芥的参考基因组为例,基因组命名为”Athaliana.fa”
第一步:为RepeatModeler创建索引数据库
1 | BuildDatabase -name ath -engine ncbi Athaliana.fa |
第二步:运行RepeatModeler从头预测
1 | RepeatModeler -database ath -engine ncbi -pa 20 &> ath.out & |
这一步运行时间相对比较久,和线程数有关。运行中的的文件存放在RM_.xxx文件夹下
1 | RM_100741.WedSep181006282019 |
运行结束后,就得到了ath-families.fa和ath-families.stk。 前者是找到的重复序列,后者是Stockholm格式的种子联配文件(seed alignment file), 可以用util/dfamConsensusTool.pl上传到Dfam_consensus数据库中。
ath-families.fa的fasta的序列部分格式为>repeatname#class/subclass,用于表明每个重复序列的归类。
第三步:根据自定义的重复序列数据库注释基因组
1 | RepeatMasker -lib ath-families.fa -e ncbi -dir . Athaliana.fa |
RepeatMasker比较常用的参数如下
-e: 搜索引擎,默认都选择ncbi-pa: 并行计算,多线程-s,-q,-qq: 搜索速度,速度和敏感度成反比-lib: 自定义重复数据库-species: 指定物种,例如human, mouse, arabidopsis-gff: 额外输出GFF文件
输出结果中, 以.masked结尾的是用N屏蔽后的序列,以tal结尾的则是统计各种重复序列的比例。