使用wgd进行全基因组复制分析
因为全基因组复制(Whole genome duplications, WGD)是生物进化的重要因素之一, 所以WGD分析也是基因组分析经常用到的一种分析方法。举个例子,我们之所以能在多条染色体之间发现一些古老基因连锁现象,是因为被子植物在过去2亿年时间里就出现了多次的全基因组复制和基因丢失事件(见下图,Tang et al., 2008)

古老WGD检测有两种方法,一种是共线性分析,另一种则是根据Ks分布图。其中Ks定义为平均每个同义位点上的同义置换数,与其对应的还有一个Ka,指的是平均每个非同义位点上的非同义置换数。
如果没有WGD或是大片段重复,那么基因组中的旁系同源基因的同义置换符合指数分布(exponential distribution), 反之,Ks分布图中就会出现一个由于WGD导致的正态分布峰(normal distributed peak). 而古老WGD的年龄则可通过分析这些峰中的同源置换数目来预测(Tiley et al., 2018)。
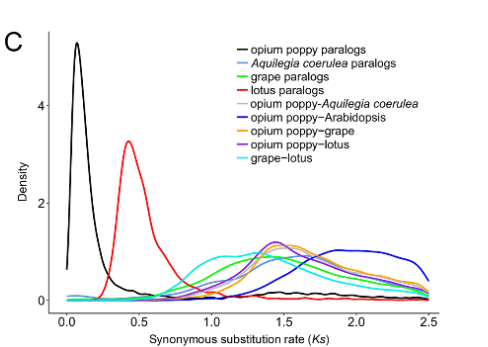
以发表在Science上的_Papaver somniferum_ L. 基因组文章中的图Fig 1C为例,文章分别分析了_P. somniferum_ 和其他几个物种的Ks分布。从Ks分布图可以看到_P. somniferum_经历了一次比较近的WGD事件(Guo et al., 2018)。
文章中关于WGD的分析流程参考附录8.1 Whole genome duplication in opium poppy genome, 我总结了关键的几点
- 使用megablast进行自比对,寻找基因组中共线性的区块
- 使用BLASTP基于RBH( reciprocal best hits )进行蛋白之间的相互比对
- 使用KaKs_Calculator基于YN模型计算RBH基因对中的Ks(synonymous substitution rate)
- 为了区分Ks中peak是WGD事件还是小规模重复,作者使用MCScanX进行共线性分析,发现93.9%的RBH基因都是基因组内共线性
目前WGD的分析流程也有人发了文章,我通过关键字”wgd pipeline”搜索找到了如下几个:
- GenoDup: https://github.com/MaoYafei/GenoDup-Pipeline
- WGDdetector: https://github.com/yongzhiyang2012/WGDdetector
- wgd: https://github.com/arzwa/wgd
这一篇介绍的是wgd的用法。
软件安装
wgd目前无法用bioconda直接安装,所以安装起来稍显麻烦,这是因为它的依赖软件有点多。wgd依赖于以下软件
- BLAST
- MCL
- MUSCLE/MAFFT/PRANK
- PAML
- PhyML/FastTree
- i-ADHoRe
但是好消息是它依赖的软件大部分都可以用bioconda进行安装
1 | conda create -n wgd python=3.5 blast mcl muscle mafft prank paml fasttree cmake libpng mpi=1.0=mpich |
这里选择了mpi=1.0=mpich, 原因是i-adhore依赖于mpich. 如果安装了openmpi就会出现error while loading shared libraries: libmpi_cxx.so.40: cannot open shared object file: No such file or directory
之后的安装就简单多了
1 | git clone https://github.com/arzwa/wgd.git |
对于i-ADHoRe,需要先在http://bioinformatics.psb.ugent.be/webtools/i-adhore/licensing/同意许可,才能下载i-ADHoRe-3.0
由于我的miniconda3安装在~/opt/下,所以安装路径为~/opt/miniconda3/envs/wgd/
1 | tar -zxvf i-adhore-3.0.01.tar.gz |
软件介绍
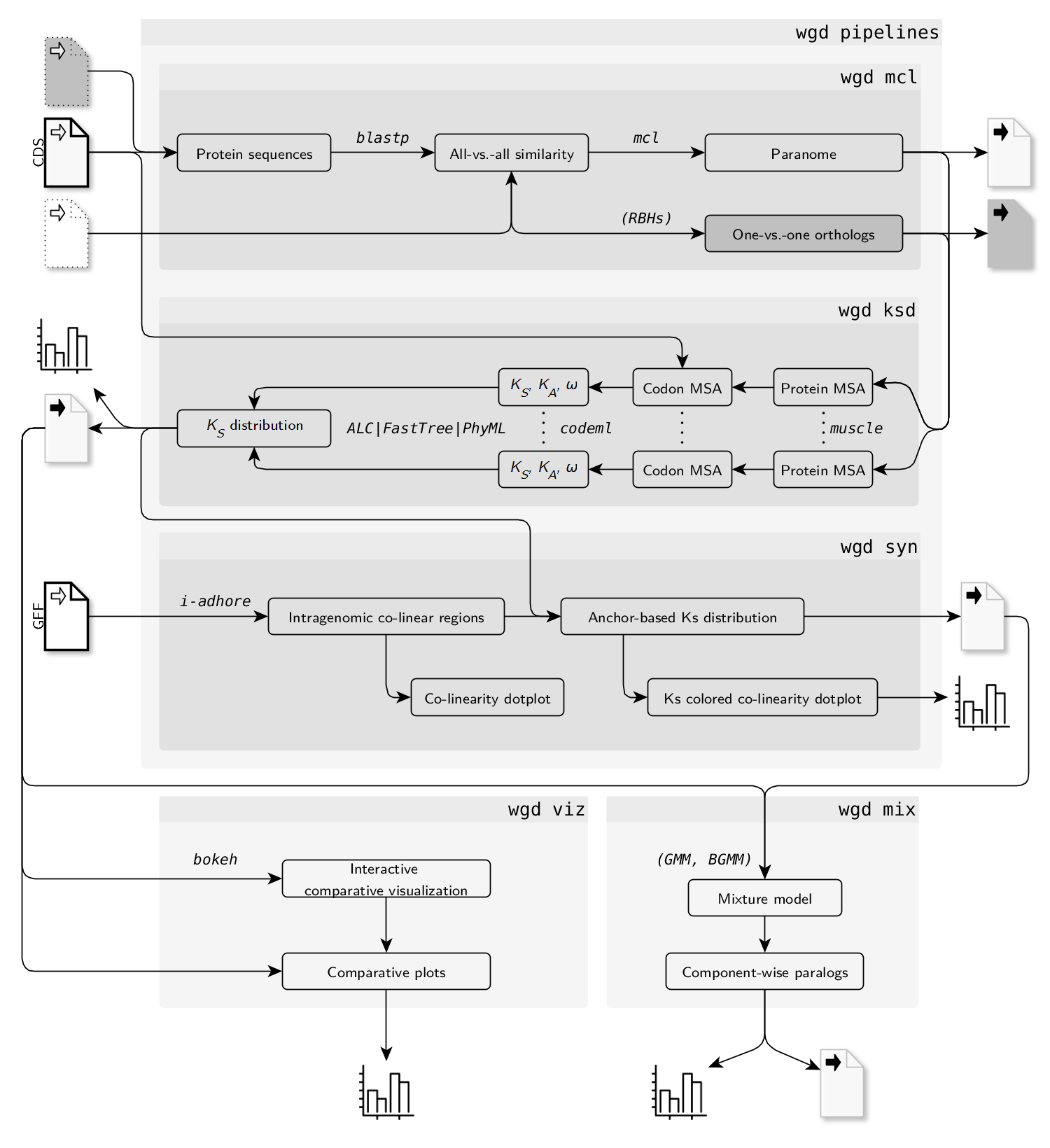
WGD一共有9个子模块
- kde: 对Ks分布进行KDE拟合
- ksd : Ks分布构建
- mcl:All-vs-ALl的BLASP比对 + MCL分类分析.
- mix: Ks分布的混合建模.
- pre: 对CDS文件进行预处理
- syn: 调用I-ADHoRe 3.0利用GFF文件进行共线性分析
- viz: 绘制柱状图和密度图
- wf1: 全基因组旁系同源基因组(paranome)的Ks标准分析流程,调用mcl, ksd和syn
- wf2: one-vs-one 同源基因(ortholog)的Ks标准分析流程,调用wcl和ksd
流程示意图如下:
使用方法
以甘蔗的基因组 Saccharum spontaneum L 为例,基因组为8倍体,共32条染色体(2n = 4x8 = 32)
下载CDS和GFF注释文件 tutorial
1 | mkdir -p wgd_tutorial && cd wgd_tutorial |
先用conda activate wgd启动我们的分析环境,然后就开始分析了
第一步: 使用wgd mcl鉴定基因组内的同源基因
1 | wgd mcl -n 20 --cds --mcl -s Sspon.v20190103.cds.fasta -o Sspon_cds.out |
这一步运行过程中,wgd会先检查cds序列是否有效,也就是是否以ATG(起始密码子)开头,且以TAA/TAG/TGA(终止密码子)结尾,然后将cds转成氨基酸序列,之后用Blastp进行相互比对,然后根据blastp结果用mcl聚类的方式寻找旁系同源基因。
输出结果在Sspon_cds.out,有两个结果输出
- blast.tsv: BLASTP的outfmt6输出结果
- blast.tsv.mcl: MCL聚类结果,每一行可以认为是一个基因家族(gene family)
第二步: 使用wgd ksd构建Ks分布
1 | wgd ksd --n_threads 80 Sspon_cds.out/Sspon.v20190103.cds.fasta.blast.tsv.mcl Sspon.v20190103.cds.fasta |
这一步也是先过滤cds中的无效数据,然后用mafft(默认)/muscle/prank对每个基因家族进行多重序列联配,用codeml计算dN/dS, 用alc/fasttree(默认)/phyml建树
输出结果在wgd_ksd目录下
- ks.tsv: 每个基因家族中基因对的各项统计,其中包括Ka和Ks
- ks.svg: Ks分布,见下图

第三步: 如果基因组质量不错,那么可以使用wgd syn进行共线性分析。它能帮我们找到基因组内的共线性区块,以及相应的锚定点
1 | wgd syn --feature gene --gene_attribute ID \ |
输出图片以.svg结尾,如下所示,图中颜色红蓝代表Ks得分。

Ks的下游分析通常还包括对Ks分布的统计建模,这可以使用wgd kde进行核密度拟合(Kernel density estimate, KDE)或用wgd mix建立高斯混合模型(Gaussian mixture models)
1 | # KDE |
wgd kde输出kde.svg, 而wdg mix则生成一个 wgd_mix文件夹。
混合模型常用于基于Ks分布研究WGD。基于一些基本的分子进化假设,我们预期Ks分布中由WGD导致的peak应该符合高斯分布,能够用log-normal分布近似。但是考虑到混合模型容易过拟合,特别是Ks分布,因此作者不建议使用混合模型作为多次WGD假设的正式统计测试。
参考文献
Tang, H., Bowers, J.E., Wang, X., Ming, R., Alam, M., and Paterson, A.H. (2008). Synteny and Collinearity in Plant Genomes. Science 320, 486–488.
Tiley, G.P., Barker, M.S., and Burleigh, J.G. (2018). Assessing the Performance of Ks Plots for Detecting Ancient Whole Genome Duplications. Genome Biol Evol 10, 2882–2898.
Guo, L., Winzer, T., Yang, X., Li, Y., Ning, Z., He, Z., Teodor, R., Lu, Y., Bowser, T.A., Graham, I.A., et al. (2018). The opium poppy genome and morphinan production. Science 362, 343–347.
Zwaenepoel, A., and Van de Peer, Y. (2019). wgd—simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics 35, 2153–2155.