Basic local alignment search tool (BLAST)
包括:blastn, blastp, blastx, tblastn, tblastx等. 使用conda安装即可。
1 | conda install -c bioconda blast |
BLAST的主要理念
- Search may take place in nucleotide and/or protein space or translated spaces where nucleotides are translated into proteins.
- Searches may implement search “strategies”: optimizations to a certain task. Different search strategies will return different alignments.
- Searches use alignments that rely on scoring matrices
- Searches may be customized with many additional parameters. BLAST has many subtle functions that most users never need.
本地BLAST的基本步骤
- 用makeblastdb为BLAST提供数据库
- 选择blast工具,blastn,blastp
- 运行工具,有必要的还可以对输出结果进行修饰
第一步:建立检索所需数据库
BLAST数据库分为两类,核酸数据库和氨基酸数据库,可以用makeblastbd创建。可以用help参数简单看下说明。
1 | makeblastdb -help |
具体以拟南芥基因组作为案例,介绍使用方法:
注: 拟南芥的基因组可以在TAIR上下在,也可在ensemblgenomes下载。后者还可以下载其他植物的基因组
1 | 下载基因组 |
如果你从NCBI或者其他渠道下载了格式化过的数据库,那么可以用blastdbcmd去检索blast数据库,参数很多,常用就如下几个
- db string : string表示数据库所在路径
- dbtype string,: string在(guess, nucl, prot)中选择一个
- 检索相关参数
- -entry all 或 555, AC147927 或 gnl|dbname|tag’
- -entry_batch 提供一个包含多个检索关键字的文件
- -info 数据库基本信息
- 输出格式 -outfmt %f %s %a %g …默认是%f
- out 输出文件
- show_blastdb_search_path: blast检索数据库路径
使用案例
1 | 查看信息 |
还有其他有意思的参数,可以看帮助文件了解
可选:BLAST安装和更新nr和nt库
安装nt/nr库需要先进行环境变量配置,在家目录下新建一个.ncbirc配置文件,然后添加如下内容
1 | ; 开始配置BLAST |
配置好之后,使用BLAST+自带的update_blastdb.pl脚本下载nr和nt等库文件(不建议下载序列文件,一是因为后者文件更大,二是因为可以从库文件中提取序列blastdbcmd -db nr -dbtype prot -entry all -outfmt “%f” -out nr.fa ,最主要是建库需要花费很长时间),直接运行下列命令即可自动下载。
1 | nohup time update_blastdb.pl nt nr > log & |
如果你不像通过update_blastdb.pl下载nr和nt等库文件,也可以是从ncbi上直接下载一系列nt/nr.xx.tar.gz文件,然后解压缩即可,后续还可以用update_blastdb.pl进行数据更新。
报错: 使用update_blastdb.pl更新和下载数据库时候出现模块未安装的问题。解决方法,首先用conda安装对应的模块,然后修改update_blastdb.pl的第一行,即shebang部分,以conda的perl替换,或者按照如下方法执行。
1 | perl `which update_blastdb.pl` |
下载过程中请确保网络状态良好,否则会出现Downloading nt.00.tar.gz...Unable to close datastream报错。
第二步:选择blast工具
根据不同的需求,比如说你用的序列是氨基酸还是核苷酸,你要查找的数据是核甘酸还是氨基酸,选择合适的blast工具。不同需求的对应关系可以见下图(来自biostars handbook)
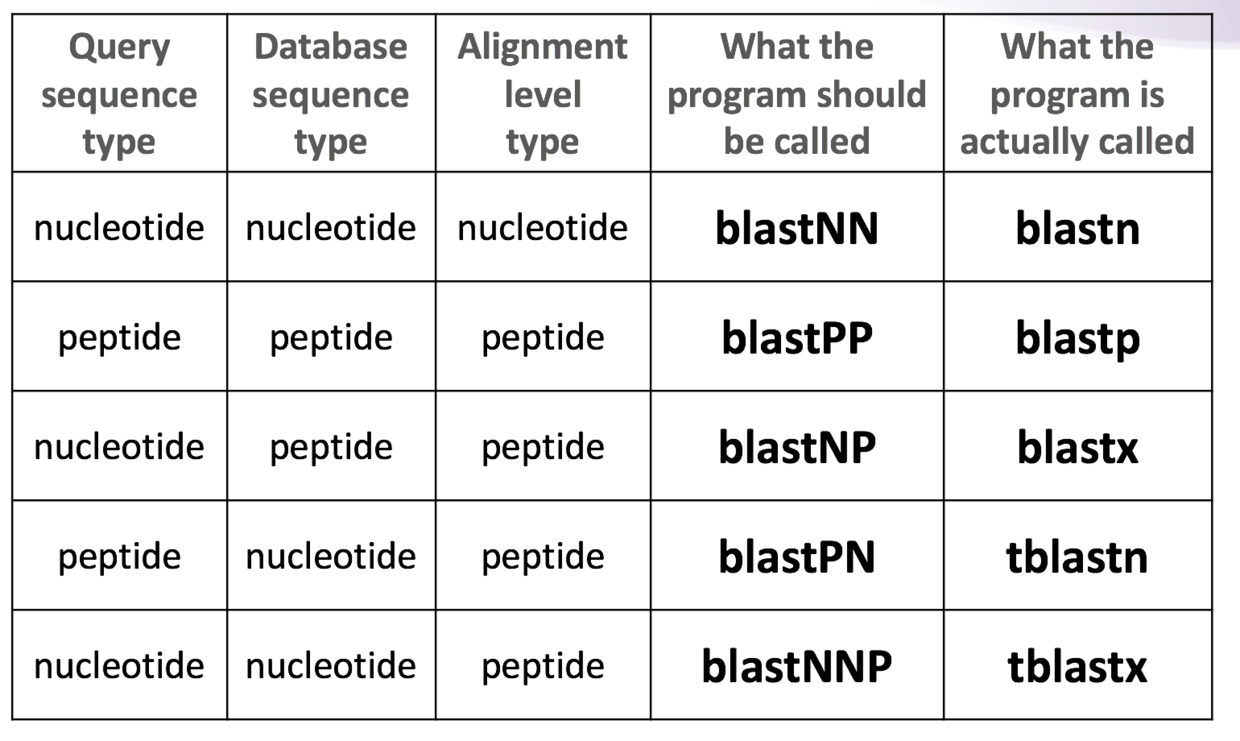
不同工具的应用范围虽然不同,但是基本参数都是一致的,学会blastn,基本上其他诸如blastp,blastx也都会了。
blastn的使用参数很多 blastn [-h] ,但是比较常用是如下几个
- -db : 数据库在本地的位置,或者是NCBI上数据库的类型,如 -db nr
-query: 检索文件
-query_loc : 指定检索的位置
-strand: 搜索正义链还是反义链,还是都要
out : 输出文件
-remote: 可以用NCBI的远程数据库, 一般与 -db nr
-evalue 科学计数法,比如说1e3,定义期望值阈值。E值表明在随机的情况下,其它序列与目标序列相似度要大于这条显示的序列的可能性。 与S值有关,S值表示两序列的同源性,分值越高表明它们之间相似的程度越大
E值总结: 1.E值适合于有一定长度,而且复杂度不能太低的序列。2. 当E值小于10-5时,表明两序列有较高的同源性,而不是因为计算错误。3. 当E值小于10-6时,表时两序列的同源性非常高,几乎没有必要再做确认。
与联配计分相关参数: -gapopen,打开gap的代价;-gapextend, gap延伸的代价;-penalty:核酸错配的惩罚; -reward, 核酸正确匹配的奖励;
结果过滤:-perc_identity, 根据相似度
注 BLAST还提供一个task参数,感觉很有用的样子,好像会针对任务进行优化速度。
第三步:运行blast,调整输出格式。
我随机找了一段序列进行检索
1 | echo '>test' > query.fa |
用的是blastn 检索核酸数据库。最简单的用法就是提供数据库所在位置和需要检索的序列文件
1 | blastn -db BLAST/TAIR10 -query query.fa |
以上是默认输出,blast的-outfmt选项提供个性化的选择。一共有18个选择,默认是0。
0 = Pairwise,
1 = Query-anchored showing identities,
2 = Query-anchored no identities,
3 = Flat query-anchored showing identities,
4 = Flat query-anchored no identities,
5 = BLAST XML,
6 = Tabular,
7 = Tabular with comment lines,
8 = Seqalign (Text ASN.1),
9 = Seqalign (Binary ASN.1),
10 = Comma-separated values,
11 = BLAST archive (ASN.1),
12 = Seqalign (JSON),
13 = Multiple-file BLAST JSON,
14 = Multiple-file BLAST XML2,
15 = Single-file BLAST JSON,
16 = Single-file BLAST XML2,
17 = Sequence Alignment/Map (SAM),
18 = Organism Report
其中6,7,10,17可以自定输出格式。默认是
qaccver saccver pident length mismatch gapopen qstart qend sstart send evalue bitscore
| 简写 | 含义 |
|---|---|
| qaccver | 查询的AC版本(与此类似的还有qseqid,qgi,qacc,与序列命名有关) |
| saccver | 目标的AC版本(于此类似的还有sseqid,sallseqid,sgi,sacc,sallacc,也是序列命名相关) |
| pident | 完全匹配百分比 (响应的nident则是匹配数) |
| length | 联配长度(另外slen表示查询序列总长度,qlen表示目标序列总长度) |
| mismatch | 错配数目 |
| gapopen | gap的数目 |
| qstart | 查询序列起始 |
| qstart | 查询序列结束 |
| sstart | 目标序列起始 |
| send | 目标序列结束 |
| evalue | 期望值 |
| bitscore | Bit得分 |
| score | 原始得分 |
| AC: | accession |
以格式7为实例进行输出,并且对在线数据库进行检索
1 | blastn -task blastn -remote -db nr -query query.fa -outfmt 7 -out query.txt |
如果想输入序列,增加对应的格式qseq, sseq
1 | blastn -query query.fa -db nr -outfmt ' 6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send qseq sseq evalue bitscore' |
对已有序列进行注释时常见的best hit only模式命令行
1 | blastn -query gene.fa -out gene.blast.txt -task megablast -db nt -num_threads 12 -evalue 1e-10 -best_hit_score_edge 0.05 -best_hit_overhang 0.25 -outfmt "7 std stitle" -perc_identity 50 -max_target_seqs 1 |
仅仅看参数依旧无用,还需要知道BLAST的Best-Hits的过滤算法。假设一个序列存在两个match结果,A和B,无论A还是B,他们的HSP(High-scoring Segment Pair, 没有gap时的最高联配得分)一定要高于best_hit_overhang,否则被过滤。如果满足下列条件则保留A
- evalue(A) >= evalue (B)
- score(A)/length(A) < (1.0-score_edge)*score(B)/length(B)
搭建网页BLAST
曾经的BLAST安装后提供wwwblast用于构建本地的BLAST网页工具,但是BLAST+没有提供这个工具,好在BLAST足够出名,也就有人给它开发网页版工具。如viroBLAST和Sequenceserver, 目前来看似乎后者更受人欢迎。
有root安装起来非常的容易
1 | sudo apt install ruby ncbi-blast+ ruby-dev rubygems-integration npm |
数据库准备,前面步骤已经下载了拟南芥基因组的FASTA格式数据
1 | sequenceserver -d /到/之前/建立/BLAST/文件路径 |
然后就可以打开浏览器输入IP:端口号使用了。
Sequenceserver高级用法
开机启动:
新建一个/etc/systemd/system/sequenceserver.service文件,添加如下内容。注意修改ExecStart.
1 | [Unit] |
然后重新加载systemctl
1 | ## let systemd know about changed files |
nginx反向代理:我承认没有基本的nginx的知识根本搞不定这一步,所以我建议组内使用就不要折腾这个。简单的说就是在nginx的配置文件的server部分添加如下内容即可。
1 | location /sequenceserver/ { |
假如你有docker的话
1 | # ubuntu |
注意,你得在宿主机器上 /path-to-database-dir建好blast索引,或者用docker exec -it docker容器名 /bin/bash 进入到容器里,用sequenceserver -d db &运行
参考资料:http://www.sequenceserver.com/doc/