有一天我的师弟提了一个需求:
对于ChIP的下游分析,我很喜欢DiffBind做差异分析然后用ChIPseeker做注释这一套流程(因为ChIPseeker的输入格式是GRange格式,而DiffBind的dba.report输出也恰好是GRange格式,两者可以无缝衔接)。我之前的套路,一直都是无脑输出所有的DiffBind结果,然后放入ChIPSeeker里面做注释,完了之后转成data.frame,然后对data.frame做subset,做后续的GO分析()。但今天突然想对ChIPseeker的注释结果直接做subset,然后分别对上下调的结果画plotAnnoPie等ChipSeeker内置的画图函数。但发现subset无法对ChipSeeker annotatePeak函数的输出结果做筛选。
当然,对于DiffBind的结果用subset,分别提取上下调,然后再注释也是可以的,不过这样感觉更麻烦。
面对需求,如果无法解决提出需求的人,那么就只能写代码实现这个需求了。于是经过一个下午的努力,我给ChIPseeker加上了subset的功能。
先来介绍下如何使用。我们需要用devtools安装最新的ChIPseeker
1
2
3
4
5
| BiocManager::install("TxDb.Hsapiens.UCSC.hg19.knownGene")
install.packages("ggupset")
install.packages("ggimage")
devtools::install_github("YuLab-SMU/ChIPseeker")
library(ChIPseeker)
|
我们以测试数据集来演示
1
2
3
4
5
| library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
peakfile <- system.file("extdata", "sample_peaks.txt", package="ChIPseeker")
peakAnno <- annotatePeak(peakfile, tssRegion=c(-3000, 3000), TxDb=txdb)
peakAnno
|
此时会输出peakAnno里的描述统计信息
1
2
3
4
5
6
7
8
9
10
11
12
13
14
| Annotated peaks generated by ChIPseeker
150/150 peaks were annotated
Genomic Annotation Summary:
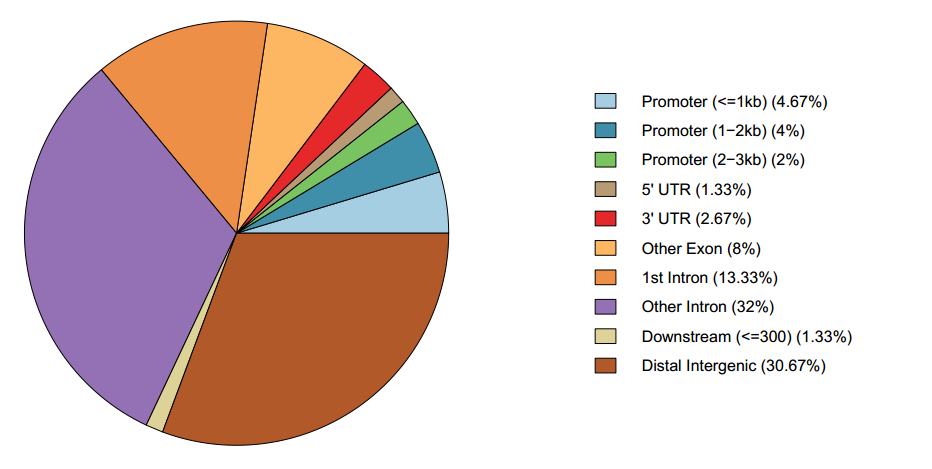
Feature Frequency
8 Promoter (<=1kb) 4.666667
9 Promoter (1-2kb) 4.000000
10 Promoter (2-3kb) 2.000000
3 5' UTR 1.333333
2 3' UTR 2.666667
6 Other Exon 8.000000
1 1st Intron 13.333333
7 Other Intron 32.000000
5 Downstream (<=300) 1.333333
4 Distal Intergenic 30.666667
|
peakAnno可以直接用来画图
1
2
| plotAnnoPie(peakAnno)
upsetplot(peakAnno)
|
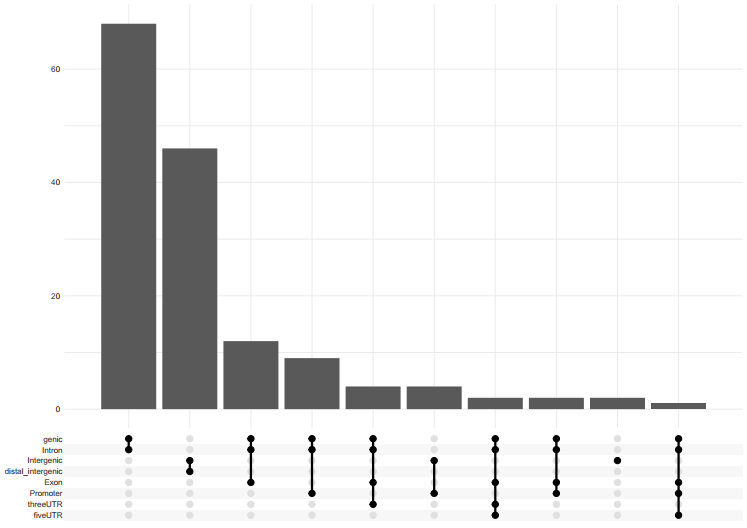
假如你突发奇想,我能不能就看看某一条染色体的作图结果,或者对于MACS2的结果,想根据FDR筛选下peak,然后看下peak的变化。
在之前我们无法直接对peakAnno背后的csAnno对象进行操作,所以需要先对输入的GRanges进行过滤然后再用annotatePeak进行注释,然后画图。
但是现在我们有了subset, 这一切就变得稍微简单起来了
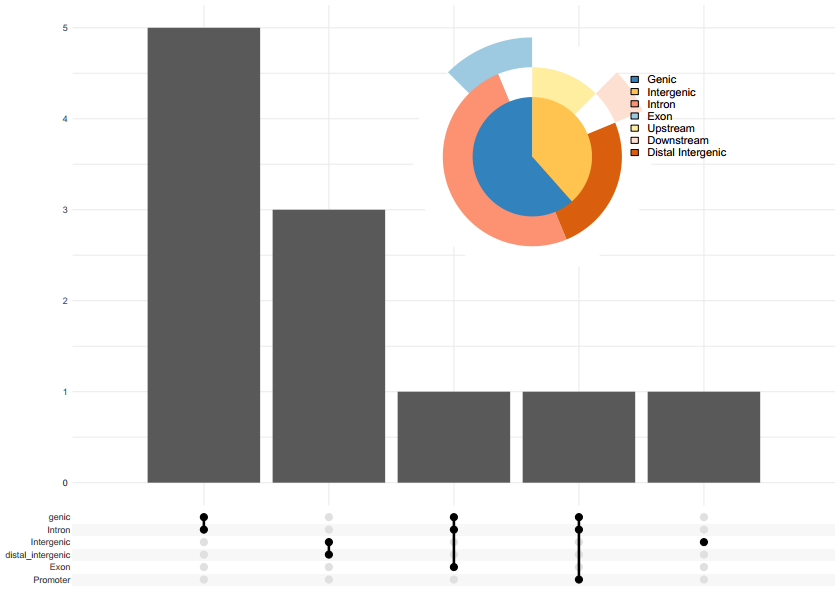
我们可以按照染色体编号进行筛选
1
2
| peakAnno_subset <- subset(peakAnno, seqnames == "chr1")
upsetplot(peakAnno_subset, vennpie = TRUE)
|
可以按照peak宽度进行筛选
1
2
| peakAnno_subset <- subset(peakAnno, length > 500)
upsetplot(peakAnno_subset, vennpie = TRUE)
|
如果筛选之后没有任何结果,那么就会报错
1
2
3
| > subset(peakAnno, FDR... < .05)
Error in names(x) <- value :
'names' attribute [2] must be the same length as the vector [1]
|
那么问题就是,这些筛选条件的名字怎么获知。
只要将peakAnno转成GRanges格式,所以可以看到的列都是你可以筛选的标准。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
| gr <- as.GRanges(peakAnno)
head(gr)
GRanges object with 2 ranges and 15 metadata columns:
seqnames ranges strand | length summit tags
<Rle> <IRanges> <Rle> | <integer> <integer> <integer>
[1] chr10 105137981-105138593 * | 614 174 7
[2] chr10 42644417-42645383 * | 968 669 27
X.10.log10.pvalue. fold_enrichment FDR...
<numeric> <numeric> <numeric>
[1] 52.8 15.32 <NA>
[2] 77.15 9.13 0.79
annotation geneChr geneStart geneEnd
<character> <integer> <integer> <integer>
[1] Exon (uc001kwv.3/6877, exon 3 of 11) 10 105127724 105148822
[2] Distal Intergenic 10 42827314 42863493
geneLength geneStrand geneId transcriptId distanceToTSS
<integer> <integer> <character> <character> <numeric>
[1] 21099 1 6877 uc010qqq.2 10257
[2] 36180 2 441666 uc010qey.2 218110
|
给ChIPseeker增加subset的功能,应该是我第一次在GitHub进行pull requests操作。写这个函数,需要先去理解csAnno对象到底是什么,以及如何保证csAnno这个对象在操作前后不会发生变化。
其实这个函数挺简单的,就是下面几行
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
|
subset.csAnno <- function(x, ... ){
index <- paste(seqnames(x@anno),start(x@anno),end(x@anno), sep = "_")
x@anno <- subset(x@anno, ...)
index2 <- paste(seqnames(x@anno),start(x@anno),end(x@anno), sep = "_")
x@detailGenomicAnnotation <- x@detailGenomicAnnotation[index %in% index2,]
x@annoStat <- getGenomicAnnoStat(x@anno)
x@peakNum <- length(x@anno)
return(x)
}
|
因为有很多功能是ChIPseeker已经有了的,比如说getGenomicAnnoStat, 还有一些是S4Vectors和BiocGenerics包提供的,比如说subset, start和end。