如何使用MUMmer比对大片段序列
测序技术刚开始发展的时候,大家得到的序列都是单个基因的长度,所以一般都是逐个基因的比较,用的都是BLAST或FASTA通过逐个基因联配的方式搜索数据库。但是1999年后,越来越多的物种全基因组出现,比如说在1999年出现了_Helicobacter pylori_的第二类菌株的基因组序列,就需要研究同一物种不同品系进化过程的基因组变化,比如说基因倒置现象。传统的BLAST/FASTA就用不了,就需要用到新的工具,这就是MUMmer出现的历史背景。
那么MUMmer能用来研究什么呢?比如说细菌的不同菌株基因组中倒置现象,人和老鼠的基因组在进化上的重排现象。还有比较同一物种的不同组装结果等。MUMmer的算法基础(suffix tree)使得它的速度比BLASTZ(k-mers)快得多,但是灵敏度低,也就是检测不到比较弱的匹配,但是作者说这都是可以通过修改参数进行改善
安装
MUMmer是开源软件,因此可以通过下载源码编译的方式进行安装,同时biconda上已经有编译好的二进制版本方便用conda进行安装。目前,我个人比较推荐使用源码编译的方式进行安装。目前MUMmer已经更新到第四版,但是还在测试中,所以文章也没有发,求稳还是用3.23.
多说一句,如果在bioconda频道上搜索mummer, 会发现一个pymummer,不要以为这是mummer的源代码用python改写,它仅仅做到了通过调用系统安装的MUMmer的工具的方式运行而已,并且功能目前实在是太弱了。
1 | # MUMmer3.23 |
为了方便使用记得将软件路径加入PATH。
MUMmer使用方法
MUMmer的核心基于 Maximal exact matching 算法开发的mummer。其他工具(nucmer,promer)都是基于mummer的开发的流程。这些流程的分析策略分为三步:
- 用
mummer在两个输入中找给定长度的极大唯一匹配( Maximal exact matching ) - 然后将这些匹配区域聚类成较大不完全联配区域, 作为锚定点(anchor)
- 最后它从每个匹配外部扩展联配, 形成有gap的联配。
Maximal exact matching
MUMmer核心是基于后缀树(suffix tree)数据结构的最大匹配路径。 根据这个算法开发出来的repeat-match和exact-tandems可以从单个序列中检测重复,mummer则是用于联配两条或两条以上的序列。由于MUMmer的其他工具基本都是基于mummer开发的,于是理解mummer就变得非常重要。
概念1:suffix tree: 表示一个字符串的所有子字符串的数据结构,比如说abc的所有子字符串就是a,ab,ac,bc,abc.
概念2:Maximal Unique Match: 指的是匹配仅在两个比较序列中各出现一次
mummer: 基于后缀树(suffix tree)数据结构,能够在两条序列中有效定位极大唯一匹配(maximal unique matches),因此它比较适用于产生一组准确匹配(exact matches)以点图形式展示,或者用来锚定从而产生逐对联配(pair-wise alignments)
大部分情况下都不会直接用到mummer,所以只要知道MUMmer历经几次升级,使得mummer可以能够只找在reference和query都唯一的匹配(第一版功能),也可以找需要在reference唯一的匹配(第二版新增),甚至不在乎是否唯一的匹配(第三版新增),参数分别为-mum,-mumreference,maxmatch。
repeat-match和exact-tandems比较少用,毕竟参数也不多,似乎有其他更好的工具能用来寻找序列中的重复区。
Clustering:聚类
MUMmer的聚类算法能够比较智能地把几个独立地匹配按照顺序聚成一块。分为两种模式gaps和mgaps。这两者差别在于是否允许重排,分别用于run-mummer1,run-mummer3.
基于
gap和mgaps的输出,第四版还提供了annotate和combineMUMs两个工具增加联配信息。
联配构建工具
基于上述两个工具,作者编写了4个工作流程,方便实际使用。
nucmer: 由Perl写的流程,用于联配很相近(closely related)核酸序列。它比较适合定位和展示高度保守的DNA序列。注意,为了提高nucmer的精确性,最好把输入序列先做遮盖(mask)避免不感兴趣的序列的联配,或者修改单一性限制降低重复导致的联配数。promer:也是Perl写的流程,它以翻译后的氨基酸序列进行联配,工作原理同nucmer.run-mummer1,run-mummer3: 两者是基于cshell写的流程,用于两个序列的常规联配,和promer,nucmer类似,只不过能够自动识别序列类型。它们擅长联配相似度高的DNA序列,找到它们的不同,也就是适合找SNP或者纠错。前者用于1v1无重排,后者1v多有重排
重点介绍一下nucmer的使用。reference和query文件都需要时fasta格式,每个都可以有多条序列。
1 | nucmer [options] <reference> <query file> |
参数我将其分为五个部分,匹配算法,聚类,外延、其他和新增
匹配:
1 | --mum, --mumreference(默认), --maxmatch |
聚类:
1 | --mincluster/-c: 用于聚类的匹配最低长度,默认65 |
外延:
1 | --breaklen/-b: 在对联配两端拓展式,在终止后继续延伸的程度,默认200 |
其他:
1 | --depend: 显示依赖信息后退出 |
新增:
1 | # 在第四版新增的参数 |
运行后得到一个delta格式的文件,它的作用是记录每个联配的坐标,每个联配中的插入和缺失的距离。下面逐行进行解释
1 | /home/username/reference.fasta /home/username/query.fasta # 两个比较文件的位置 |
用法举例
两个完整度高的基因组
比较常见的用法是把一条连续的序列和另一条连续的序列比。比如说两个细菌的菌株,直接用mummer
1 | wget http://mummer.sourceforge.net/examples/data/H_pylori26695_Eslice.fasta |
或者是高度相似序列,不含重排
1 | run-mummer1 ref.fasta qry.fasta ref_qry |
或者是高度相似序列,存在重排现象
1 | run-mummer3 ref.fasta qry.fasta ref_qry |
以上的run-mummer*比较关注序列的不同之处,那么对于相似度没有那么高的两个序列,就需要用到nucmer。nucmer关注序列的相似之处,所以它允许重排,倒置和重复现象。nucmer允许多对多的比较方式,当然比较常用的是多对一的比较。
1 | nucmer --maxgap=500 --mincluster=100 --prefix=ref_qry ref.fasta qry.fasta |
注意一点: 第四版中
run-mummer1, run-mummer3已经被废弃了,就是尽管保留了,但是没有对它做任何升级的意思。
如果是有点差异的两个序列,可以用翻译的氨基酸序列进行比较
1 | promer --prefix=ref_qry ref.fasta qry.fasta |
两个基因草图
上面都是两条序列间的比较,但是研究植物的人更容易遇到的是两个物种的基因组都只有scafold级别,甚至是contig级别。那么就可以使用nucmer或promer构建序列间的可能联配。
1 | # 首先过滤低于1kb的序列 |
一个基因草图对一个完整基因组
这里可以比较一下水稻日本晴基因组和其他地方品种
1 | nucmer --prefix IRGSP1_DHX2 ~/reference/genome/IRGSP1.0/IRGSP-1.0_genome.fasta ~/reference/genome/rice_contigs/DHX2.fa |
在第四版中新增了一个
dnadiff,进一步封装nucmer和其他数据整理工具,基本上没啥参数,而输出很齐全,非常的人性化。在不知如何开始的时候,可以无脑用这个。
1 | # 已有delta文件 |
数据整理
之前得到的数据还需要用delta-filter,show-coords和show-tilling进行进一步整理才能用于后续的分析。后续操作基于上面的基因草图和完成基因组比较结果。
最初的比对结果保留了最多的信息,需要用delta-filter进行一波过滤,除去不太合适的部分。过滤选项有
-i: 最小的相似度 [0,100], 默认0-l: 最小的匹配长度 默认0.-u: 最小的联配唯一度 [0,100], 默认0-o: 最大重叠度,针对-r和-q设置。 [0,100], 默认100-g: 1对1全局匹配,不允许重排-1: 1对1联配,允许重排,是-r和-q的交集-m: 多对对联配,允许重排,是-r和-q的合集。-q: 仅保留每个query在reference上的最佳位置,允许多条query在reference上重叠-r: 仅保留每个reference在query上的最佳位置,允许多条reference在query上重叠
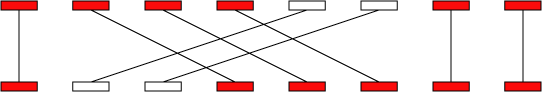
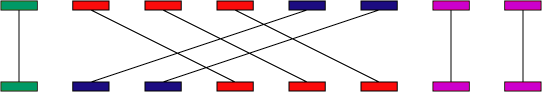
以上顺序是-i -l -u -q -r -g -m -1.光看参数估计不太明白,来一波图解。referece的一个片段可以联配到query的多个片段上,同样的query的一个片段也可以联配到reference的多个片段上,那么如何取舍呢?

通过-i,-l可以先过滤一些比较短,并且相似度比较低的匹配情况。进一步,计算长度和相似度的乘积(加权最长增加子集),对于-q而言就是保留左2,对于-r则是保留右3. 这就是传说中的三角关系,这种关系可以用-m保留或者用-q消灭。
比如说我想看contig和reference两者唯一匹配,并且长度在1000,相似度大于90.
1 | delta-filter -i 89 -l 1000 -1 IRGSP1_DHX2.delta > IRGSP1_DHX2_i89_l1000_1.delta.filter |
如何才能验证上面参数运行的结果是符合要求的呢?毕竟数据分析第一原则“不要轻易相信分析结果,需要多次验证才能使用”。
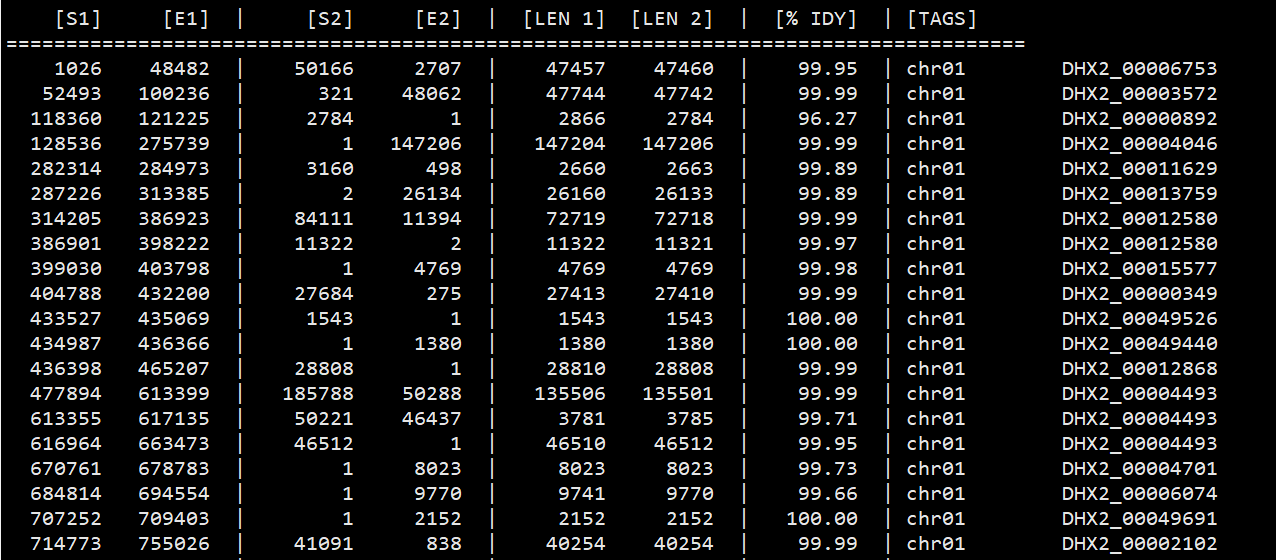
可以先用show-coord以人类可读的格式显示匹配的坐标。
1 | show-coords -r IRGSP1_DHX2_i89_l1000_1.delta.filter > IRGSP1_DHX2_i89_l1000_1.coord |
不难发现这个位置锚定的非常不错,至少暂时看起来没有重叠之处
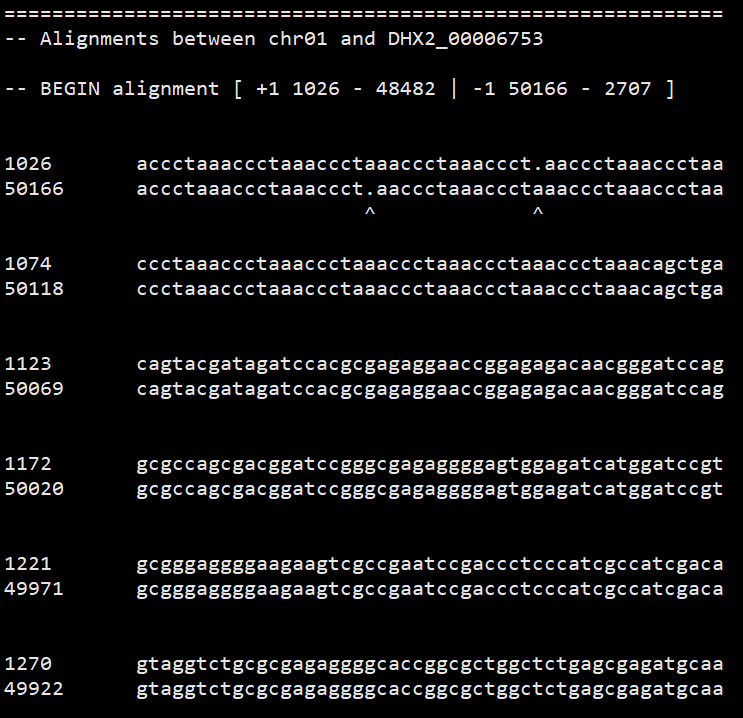
用show-aligns看某一个匹配的序列比对情况。
1 | show-aligns IRGSP1_DHX2_i89_l1000_1.delta.filter chr01 DHX2_00006753 | less |
针对reference有很长的组装序列的情况,还可以用show-tilling将contig回贴到reference上,如果装了gnuplot还能用mummerplot可视化点图.show-tiling会尝试根据contig和reference匹配信息构建出tiling path(不好翻译呀。。),不怎么用得到。
show-snps可以根据delta文件整理出SNP信息,我表示也没有怎么用到。