水稻转录组分析实战
文章标题: Genes, pathways and transcription factors involved in seedling stage chilling stress tolerance in indica rice through RNA-Seq analysis
该文章发表在BMC plant biology,植物科学2区SCI,近四年IF稳定在3分左右。这篇文章的核心部分就是测了好几个转录组,然后做了一点实验验证。
文章简介
本部分内容由我的一位师弟完成
背景
世界50%以上的人口将稻米作为饮食中碳水化合物的主要来源。这是因为水稻的种植范围很广,从山地到平原地区均有种植。正因如此,水稻的一生会面临多种生物胁迫(如病原菌,昆虫等)和非生物胁迫(如寒冷,高温,盐碱等),因此解析水稻的抗逆机制,培育具有多种抗逆性状的水稻具有很大的应用价值。而本文的作者是印度人,印度大部分地区的气候具有明显的旱雨两季,旱季的水稻在苗期易受冷(寒)害的影响。随着RNA-seq手段越来越成熟,在比较不同转录组的差异中发挥了重要作用,这对做基因功能的实验室来讲无疑是巨大的福音。作者就想利用RNA-seq的手段,寻找耐寒品种(Geetanjali)和冷敏感品种(Shahabhagidhan)在转录组水平的差异表达的基因,以期在水稻耐寒过程中鉴定到新颖的转录本,并能获得对基因不同表达水平的认识。
方法
总体思路,对耐寒和冷敏感的品种在冷处理过程中的不同时间点取样,并进行转录组分析,分析完成后,用qRT-PCR来验证表达上调和下调的基因,进一步confirm分析结果。
结果
基于低温胁迫处理的5个不同时间点的比较转录组分析显示:在冷敏感品种中有更多的下调基因,在耐寒品种中有更多上调的基因。 在冷敏感品种(CSV)中检测到总计13930个差异基因,而在耐寒品种(CTV)中检测到10599个差异基因,且随时间的延长,差异基因的数目也增加。GO富集分析揭示了18个CSV项和28个CTV项显著参与到molecular function, cellular component and biological process。GO分类揭示了耐寒过程中的差异基因显著参与转录调控,活性氧爆发,脂质结合,催化和水解酶活性的作用。KEGG注释通路揭示了更多数量的基因在耐寒过程中调节不同的通路来发挥作用。
结论
结论真的是简单到吐血,真的是简单到吐血,真的是简单到吐血。就一句话:通过对耐寒品种的转录组分析,我们检测到了涉及到抗逆过程的基因,通路和转录因子。
转录组分析重现
转录组分析环境的搭建和一个分析流程全过程可以哔哩哔哩上搜索”徐洲更”, 即可看我录制的视频。对于学习过程中出现的问题,可以参加我们的付费答疑群,活动见https://mp.weixin.qq.com/s/w47Hc2BmRCLpQMlRZ4rCCw。
本次文章重现涉及到的软件如下:
- 命令行:prefetch, fasterq-dump, hisat2, samtools, featureCounts, gffread
- R:DESeq2, clusterProfiler
1 | conda create -n rna-seq sra-tools fastqc fastp hisat2 samtools subread gffread multiqc |
数据下载
文章用到的RNA-seq数据编号,PRJNA288892, 一共有12个样本。我以这个项目编号新建一个文件夹,开始本次的分析
1 | mkdir -p PRJNA288892 && cd PRJNA288892 |
接下来是下载文章用到的数据,编号存在SRR_Acc_List.txt
1 | SRR2089751 |
建立一个sra文件夹,使用prefetch将数据下载到该文件夹下
1 | mkdir sra |
然后将sra文件转成fastq格式
1 | mkdir -p 00-raw-data |
原始数据质控
原文只说了High quality (QV > 25) 的read用于比对,并没有提到具体的质控的软件。我们这里用fastp做质控,因为fastp跑完之后也会输出一个网页,所以我也就懒得用FastQC再跑一次
1 | mkdir -p 01-clean-data |
重申一个观点,对于普通转录组,由于测序质量过关,比对软件也能处理开头的接头序列,我基本不做质控。
建立索引
文章说它用到水稻参考基因组和注释文件下载自ftp://ftp.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version_7.0/all.dir/, 而有趣的是这个链接我打不开,目前可用的下载地址其实是: http://rice.plantbiology.msu.edu/pub/data/Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_7.0/all.dir/
1 | mkdir reference && cd reference |
此外文章用的是TopHat v1.3.3和Bowtie v0.12.9 默认参数做的比对,我对此不作任何评价。我们这里用HISAT2建立索引
1 | hhisat2-build -p 10 reference/all.con reference/Osativa |
序列比对
用HISAT2将过滤后的fastq文件和参考基因组进行比对,得到下游分析所需要的BAM文件
1 | mkdir -p 02-read-align |
表达量定量
文章使用的Cufflinks v1.3.0对基因的表达量进行定量,使用FPKM进行了标准化。我这里也不做评价,我们用featureCounts进行表达量定量
先将GFF3转成GTF格式
1 | gffread reference/all.gff3 -T -o reference/Osativa.gtf |
然后用featureCoutns
1 | mkdir -p 03-gene-count |
最终的结果,就可以读取到R语言里面进行分析了
MultiQC
上面每一步的中间文件都被我放到了qc文件夹里,因此可以用multiqc进行整合
差异分析
由于前面几步的运算时间比较久,我这里直接准备了表达量矩阵,下载方式为百度网盘链接:https://pan.baidu.com/s/1c_mdGlDggV2DH79H-X7J_g 提取码:jmi5
差异表达分析用但是精确检验(exact test)。
之前的SRR编号在分析的时候最好转成实际的样本编号,方便分析的时候查看
| 编号 | 品种 | 处理 |
|---|---|---|
| SRR2089751.sra | Sahabhagidhan | 0h control(易感品种对照) |
| SRR2089753.sra | Sahabhagidhan | 6h冷害处理 |
| SRR2089754.sra | Sahabhagidhan | 12h冷害处理 |
| SRR2089756.sra | Sahabhagidhan | 24h冷害处理 |
| SRR2093937.sra | Sahabhagidhan | 48h冷害处理 |
| SRR2093938.sra | Sahabhagidhan | 48h冷害处理后又24h恢复 |
| SRR2093939.sra | Geetanjali | 0h conrol |
| SRR2093948.sra | Geetanjali | 6h冷害处理 |
| SRR2093955.sra | Geetanjali | 12h冷害处理 |
| SRR2093959.sra | Geetanjali | 24h冷害处理 |
| SRR2093960.sra | Geetanjali | 48h冷害处理 |
| SRR2093961.sra | Geetanjali | 48h冷害处理后又24h恢复 |
Sahabhagidhan是不耐冷的品种(cold susceptible variety, CSV) ,而Geetanjali则是耐冷品种 (CTV, cold tolerant variety) 。
我们这里分析的时候把两个物种在6, 12, 24, 48时间点的样本作为重复,仅仅比较不同物种在冷害处理后的差异基因,而不分析冷害处理后随时间点变化的基因。
加载数据:
1 | df <- read.table("gene_counts", header = T, |
DESeq2差异分析
1 | library(DESeq2) |
PCA比较下组内差异是不是比组间差异小
1 | rld <- rlog(dds) |
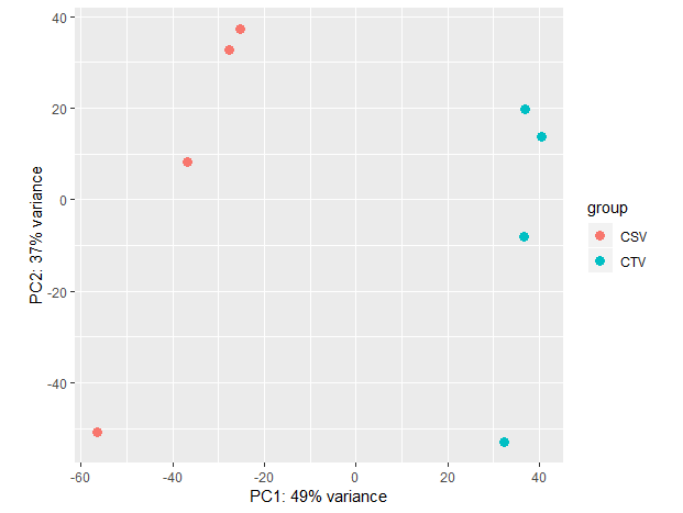
DESeq2的差异表达分析,只需要一行代码
1 | res <- results(dds) |
用ggplot2画个火山图,
1 | plot_df <- data.frame(res) |
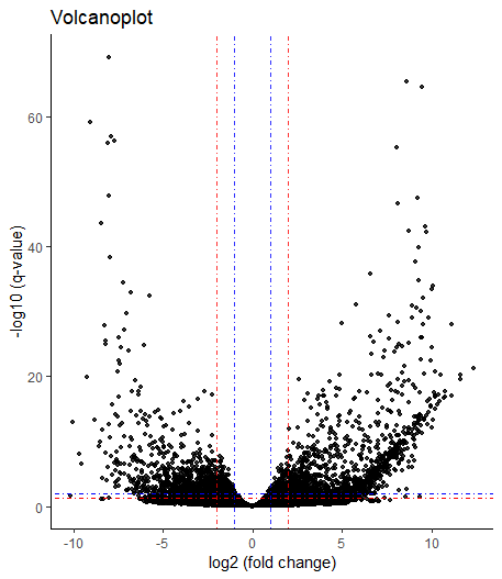
火山图的作用,一是检查你的差异分析是否合理,也就是大部分基因都是没有变化,只有一部分是差异基因,第二个是可以帮你确定阈值。
我发现即便是LogFoldChange > 2(相差4倍), p.adj < 0.01,依旧是有很多差异基因。所以我这里以这个为标准进行筛选,
1 | res_sub <- subset(res, abs(log2FoldChange) > 2 & padj <0.01) |
其实这个时候你会有一个问题,所谓的基因上调和下调是谁相对于谁呢?很简单,直接把差异的基因的标准化矩阵提取出来即可。
1 | gene_counts <- counts(dds, normalized = TRUE) |
当你看到具体的数值时
1 | CSV_1 CSV_2 CSV_3 CSV_4 CTV_1 |
你自然就知道了CTV vs CSV里,CSV是作为分母,而CTV是分子。
1 | log2 fold change (MLE): condition CTV vs CSV |
使用clusterProfiler做GO/KEGG富集分析
文章用agriGo对DEG进行分类,用singular enrichment analysis (SEA) 做GO富集分析,进一步的注释用Web Gene Ontology Annotation Plot (WEGO) 进行画图。做KEGG富集分析时,用的是KEGG GENES database KAAS (KEGG Automatic Annotation Server) 对差异基因进行功能注释.
这里我也不做评价,我们此处用Y叔的clusterProfiler做GO/KEGG富集分析。
在做GO富集分析的时候,我遇到了一个难题,我发现Bioconductor上并没有水稻的物种注释包。解决这个问题有两种方法,
- 一种是利用AnnotationHub在线检索抓取OrgDb, 但是这些包都是用ENTREZID,你需要先将RAP-DB或者MSU转为ENTREZID才行
- 另一种则是我构建好一个物种注释包,你们只要安装就可以继续使用
enrichGO, 参考https://github.com/xuzhougeng/org.Osativa.eg.db
参考我的GitHub页面安装,https://github.com/xuzhougeng/org.Osativa.eg.db
1 | install.packages("https://github.com/xuzhougeng/org.Osativa.eg.db/releases/download/v0.01/org.Osativa.eg.db.tar.gz", |
目前这个物种包比较简单,就只能做GO富集分析,以及MSU和RAP-DB之间的ID转换
然后进行GO富集分析
1 | library(clusterProfiler) |
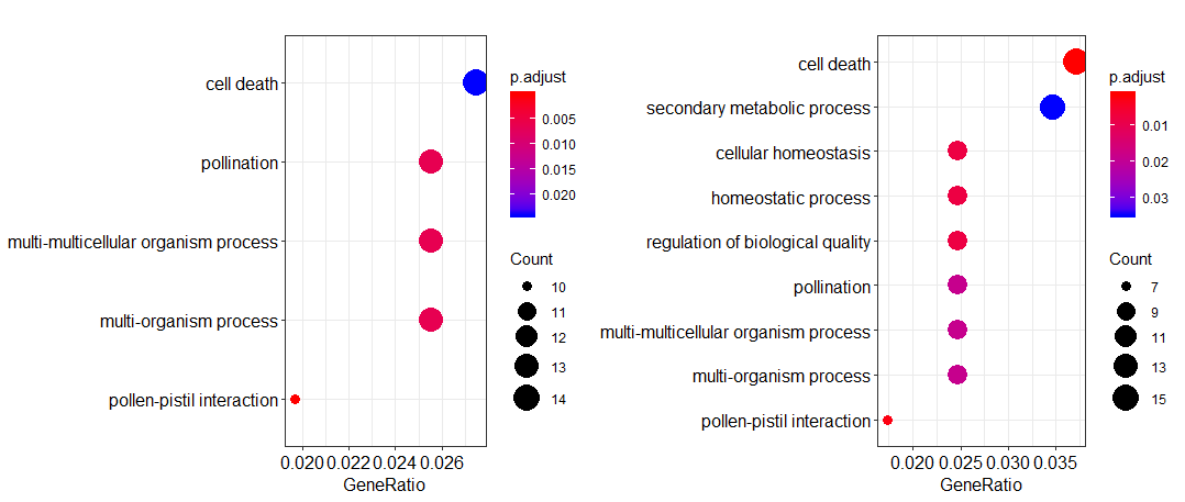
除了点图,我们还可以用clusterProfiler画下面这个图,展现每个基因在不同GO通路的倍数变化
1 | fc <- plot_df$log2FoldChange |
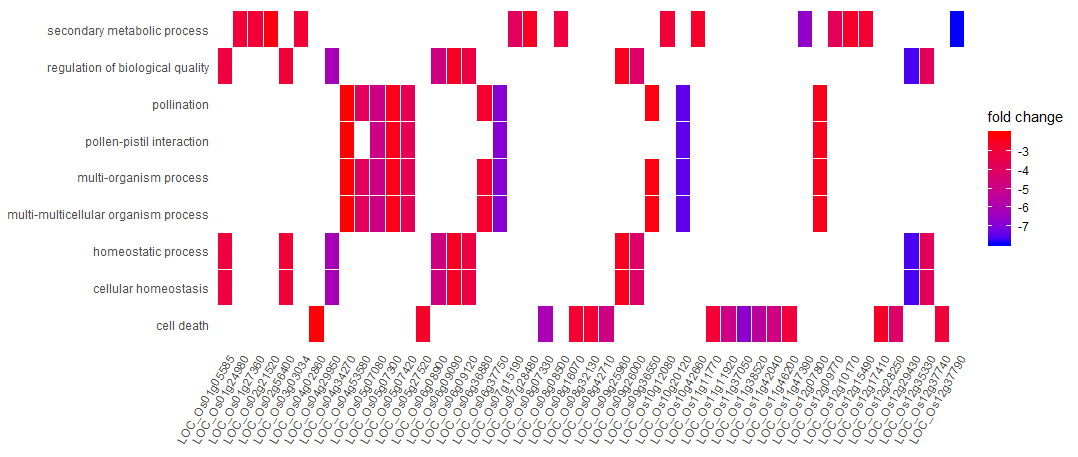
讲真,GO富集分析给的信息真的是少,我以为是我的分析出现了问题,所以我去看了文章里GO分析中BP部分,发现信息是一样的少。
GO富集分析做完之后,KEGG富集分析怎么做呢?参考我之前的这篇https://mp.weixin.qq.com/s/UnUPVoaMpfJWCQEpkmdTWA
因为KEGG要求的输入编号是RAP-DB,所以我们需要进行一次ID转换,在编号中加上”-01”,将编号中的”g”改成”t”, 就可以做KEGG富集分析了
1 | rap_id <- mapIds(x = org, |
没成想,KEGG的分析结果中只得到了一个结果, 所以也懒得放气泡图了。也难怪文章只是将差异基因按照KEGG进行了分类,而没有做富集。我这里也用Y叔的包对上调差异基因按照KEGG归类
1 | rice_kegg <- clusterProfiler::download_KEGG("dosa") |
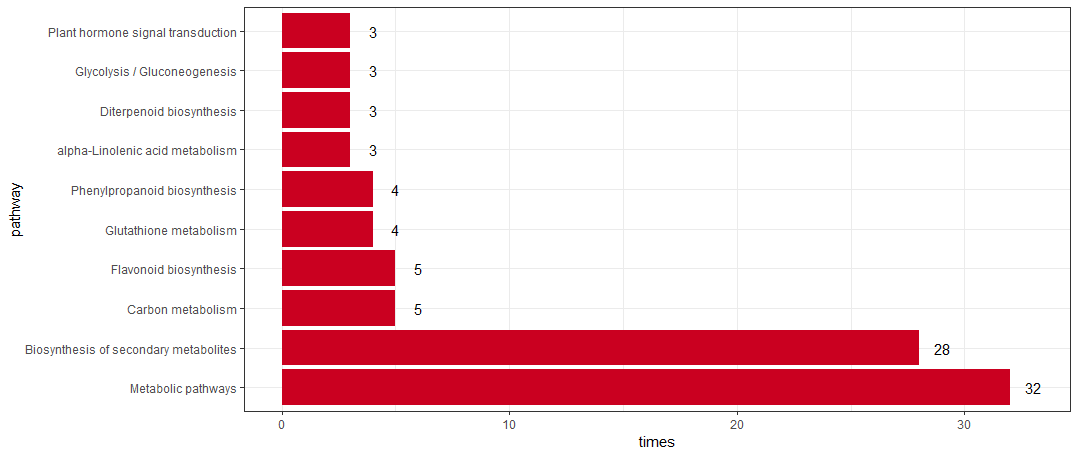
当然上下调一起展示也不难,就是代码会长一点
1 | rice_kegg <- clusterProfiler::download_KEGG("dosa") |
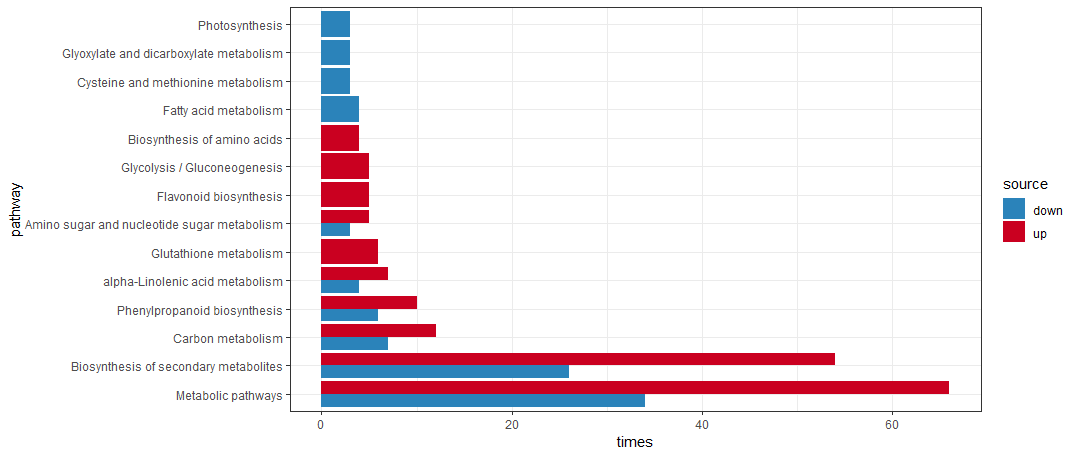
这里这展示了代码,具体如何讲故事,如何调整参数,其实就靠作者自由发挥了。
参考资料
水稻两大权威注释组织