最近(2019.8)在NBT上发表了一篇单细胞ATAC-seq的文章,题为 “Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion”。这篇文章由ATAC-seq的发明者所在的实验室用了10X Genomic 公司开发的sc-ATAC-seq技术分析而来。
文章一共测了20w个细胞,GEO编号是GSE129785, 里面是分析后的数据,而原始的FASTQ数据则在https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA532774, 数据量400G。此外,原始数据和处理过的数据还可以从https://support.10xgenomics.com/single-cell-atac/datasets进行下载。
我下载了其中66.7G的tar文件,解压后放在了百度网盘(链接:https://pan.baidu.com/s/1fu4MOhhW3aFtInsiJ5zBWg 提取码:6j7d ).
最为更重要的是,文章提供了核心分析部分用到的代码,代码地址为:https://github.com/GreenleafLab/10x-scATAC-2019
因此,我们就可以根据文章的数据和代码来学习10X sc-ATAC-seq的数据分析方法, 我们的目标是尽量重复出文章里出现的图,
文章数据
这篇文章能发到NBT,我觉得很大一部分的原因是它用大量的数据证明了scATAC-seq的可靠性,GSE129785编号中里放了67个样本,其中的GSE129785_RAW.tar(里面的文件是处理后的fragments.tsv.gz)有66.7G,解压后之后都是Cell Ranger ATAC (v 1.1.0) 处理后的fragments.gz文件, 我根据文章内容分门别类介绍这些数据:
第零类: 人类GM12878细胞系和小鼠A20细胞系(B lymphocytes)混合测序,通过滴定的方式来获取不同的细胞量,从而评估双/多细胞率(原文计算的结果是约1%)。一共有4个样本,如下
BY_500, BY_1k, BY_5k, BY_10k
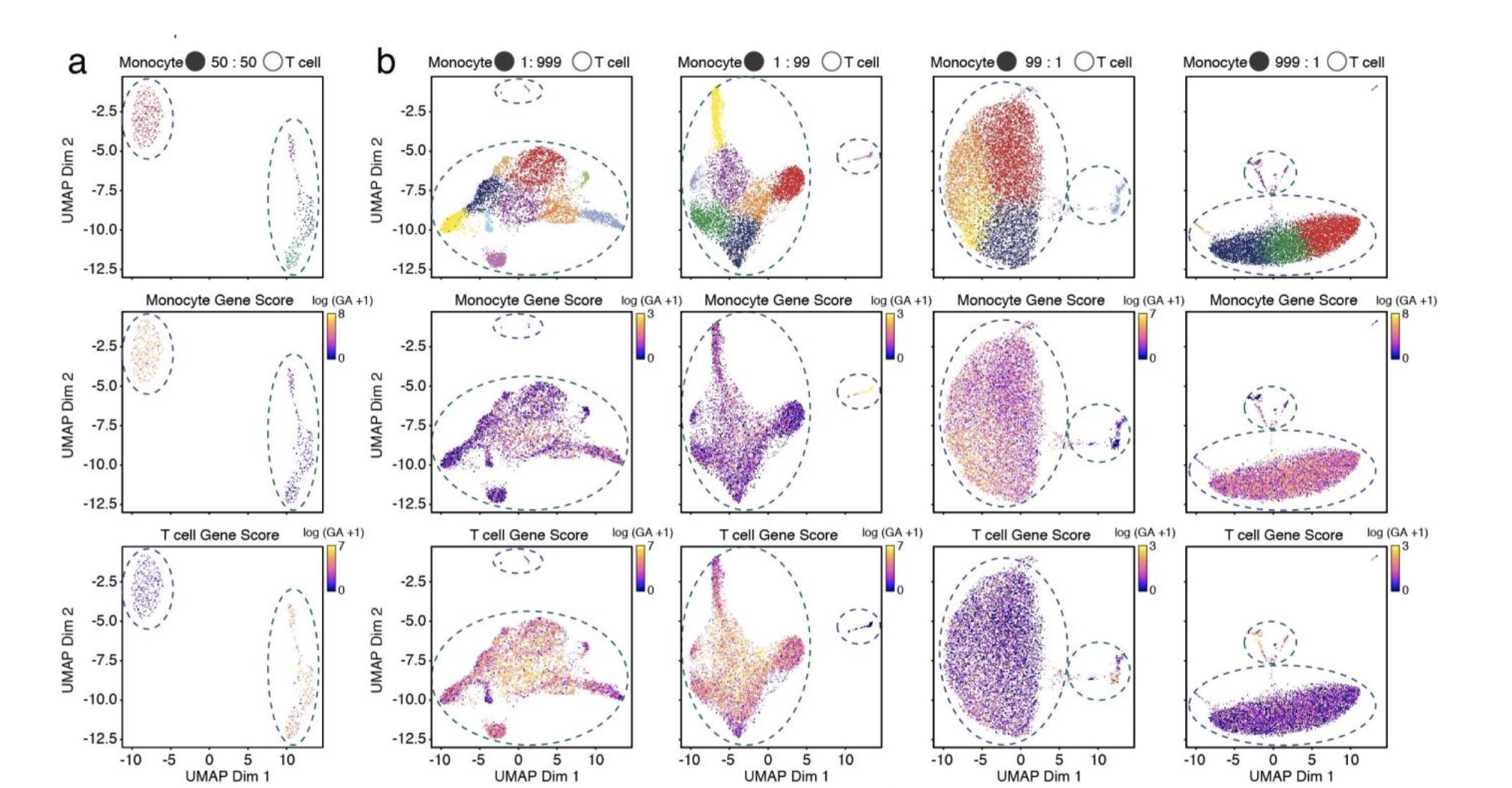
第一类:人类PMBC中不同比例的不同类型细胞混合。作者的目的是为了分析多少细胞量会被鉴定出真实的类群,作者发现,百分之一或者千分之一的比例就可以鉴定出细胞类群,有14个样本
0p1_99p9_CD4Mem_CD8Naive, 0p1_99p9_Mono_T, 0p5_99p5_CD4Mem_CD8Naive, 0p5_99p5_Mono_T, 1_99_CD4Mem_CD8Naive, 1_99_Mono_T, 50_50_CD4Mem_CD8Naive, 50_50_Mono_T, 99_1_CD4Mem_CD8Naive, 99_1_MonoT, 99p5_0p5_CD4Mem_CD8Naive, 99p5_0p5_MonoT, 99p9_0p1_CD4Mem_CD8Naive, 99p9_0p1_Mono_T
其中,p是point的简写,0p1_99p9_CD4Mem_CD8Naive就是1 CD4Mem : 999 CD8Naive. 其中MonoT是作者手误,应该是Mono_T。文章里面只提到了monocytes 和 T cells,但是这里面的数据表明还有不同比例的CD细胞混合。
第二类:fresh PBMC和viably frozen PBMC,后者分为分选和未分选。作者的目的是为了确定即便是冻存的细胞也是能够做sc-ATAC-seq。有3个样本
Fresh_pbmc_5k, Frozen_sorted_pbmc_5k, Frozen_unsorted_pbmc_5k。
第三类:16个人健康人的peripheral blood, 4个重复
PBMC_Rep1, PBMC_Rep2, PBMC_Rep3, PBMC_Rep4
第四类:16个人健康人的 bone marrow cells, 就1个样本
Bone_Marrow_Rep1
第五类:16个人健康人的富集特定表面标记的细胞,
Dendritic_Cells, Monocytes, B_Cells, CD34_Progenitors_Rep1, Regulatory_T_Cells, Naive_CD4_T_Cells_Rep1, Memory_CD4_T_Cells_Rep1, CD4_HelperT, CD4_Memory, CD4_Naive, NK_Cells, Naive_CD8_T_Cells, Memory_CD8_T_Cells, Dendritic_all_cells, CD34_Progenitors_Rep2, Memory_CD4_T_Cells_Rep2, Naive_CD4_T_Cells_Rep2
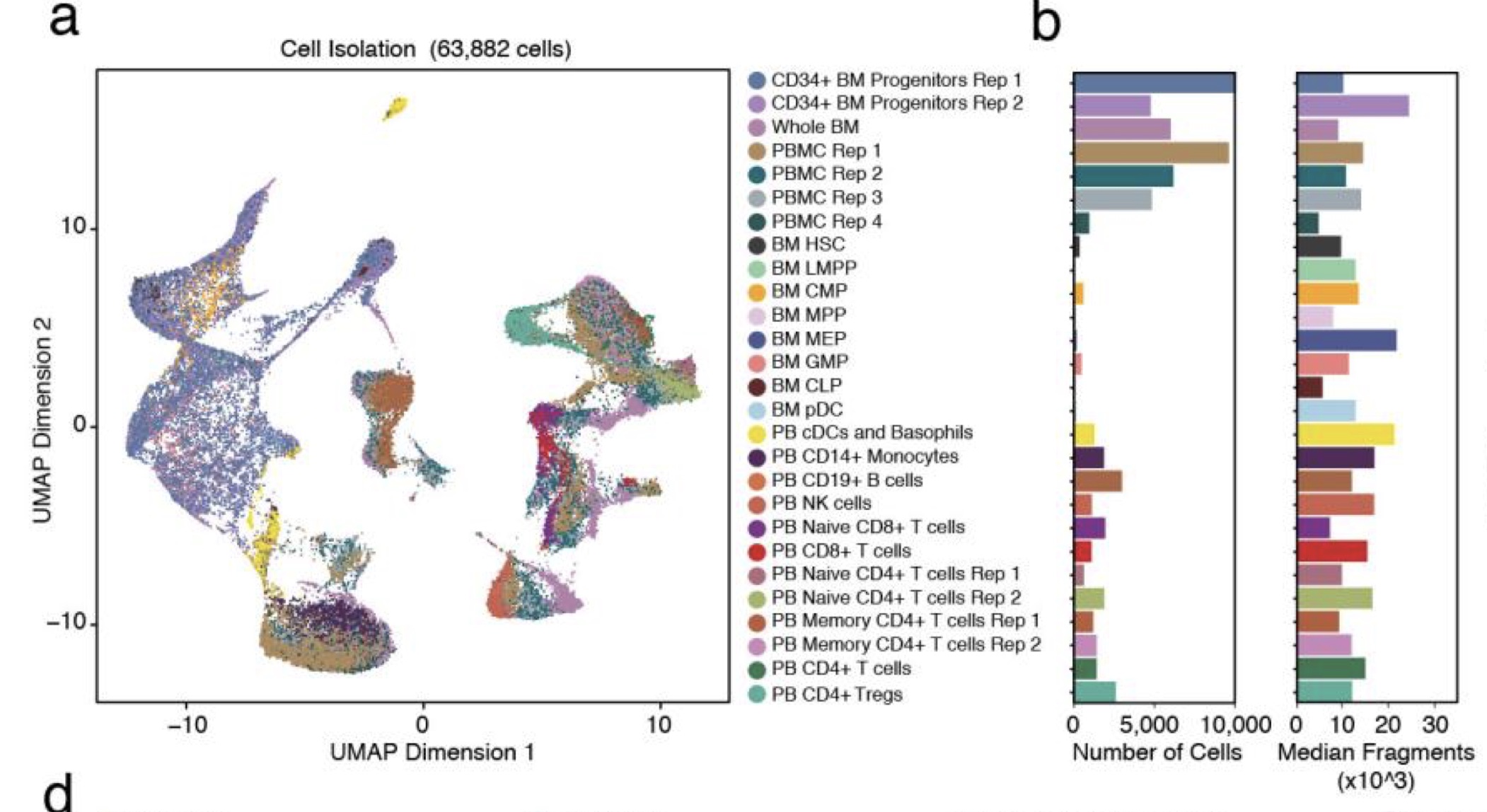
第三,四,五类其实都是健康人的免疫细胞, 文章过滤后有61,806个细胞, 聚类结果见附录sup fig. 3a
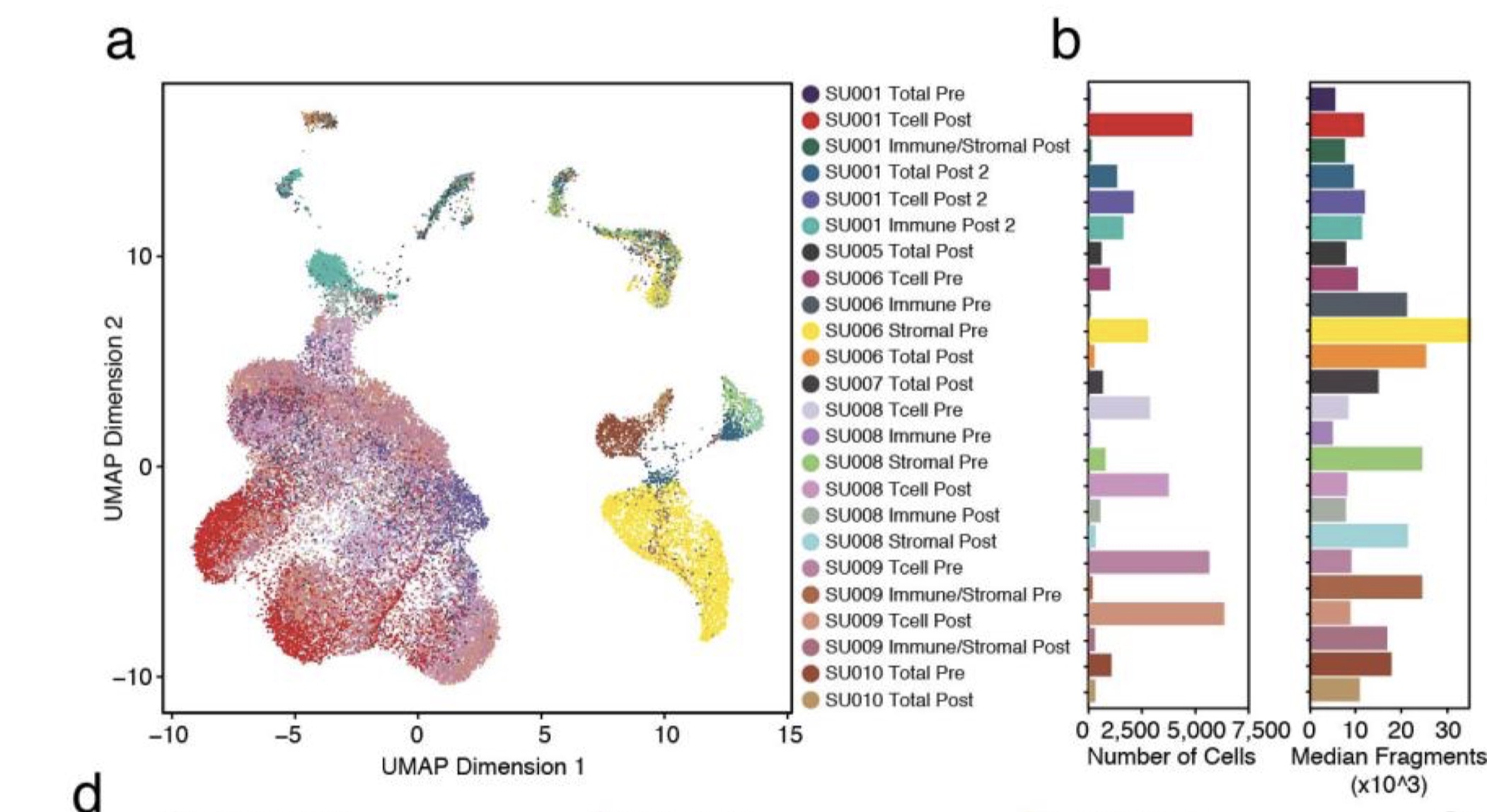
第六类:免疫疗法前后的细胞,site-matched serial tumor biopsies pre- and post-PD-1 blockade (pembrolizumab) from five patients, plus post-therapy biopsies from two additional patients, 一共有21个样本,过滤后有37,818个细胞
- SU001_Immune_Post2, SU001_Tcell_Post2, SU001_Tcell_Post, SU001_Total_Post2, SU001_Total_Pre, SU001_Tumor_Immune_Post
- SU006_Immune_Pre, SU006_Tcell_Pre, SU006_Total_Post, SU006_Tumor_Pre
- SU008_Immune_Post, SU008_Immune_Pre, SU008_Tcell_Post, SU008_Tcell_Pre, SU008_Tumor_Post, SU008_Tumor_Pre
- SU009_Tcell_Post, SU009_Tcell_Pre, SU009_Tumor_Immune_Post, SU009_Tumor_Immune_Pre
- SU010_Total_Post, SU010_Total_Pre
- SU005_Total_Post
- SU007_Total_Post
注: 这里前5个病人还可以分类,一种是直接测Total,也就是unbiased fashion,另一种区分了Immune, Tcell, Tumor_Immune, 也就是cell sorting后分开测, T cells (CD45 + CD3 + ), non-T immune cells (CD45 + CD3 − ), stromal and tumor cells (CD45 − ), 参考附录sup fig. 6a
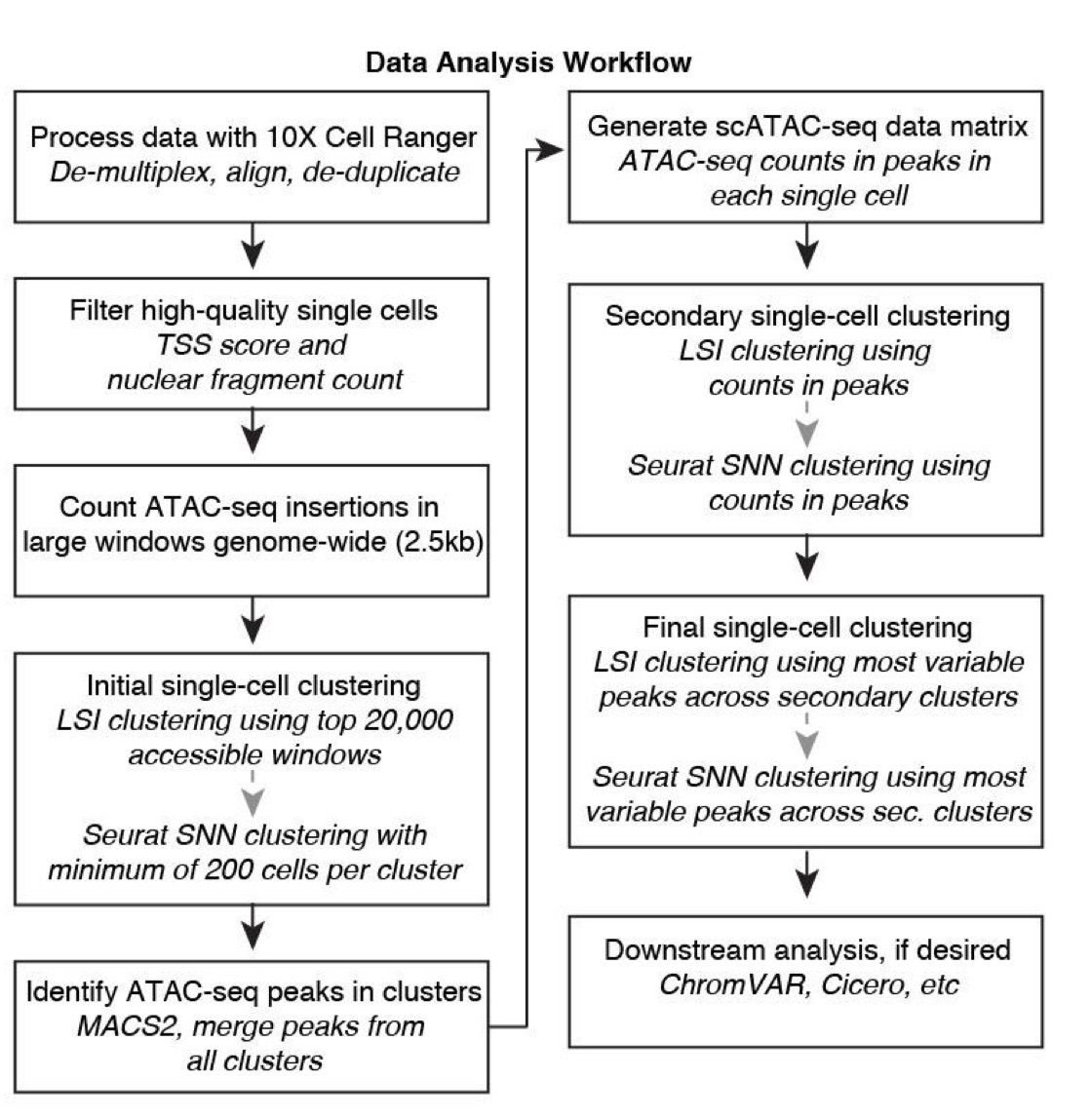
对于这些数据,文章用的是下面这套流程来进行处理
数据预处理
文章的数据预处理交给了Cell Ranger ATAC (v 1.1.0) ,用的是hg19 作为参考基因组,并不是最新GRCh38。
经@Jimmy @王诗翔 @伊现富老师 解释,是因为hg19的惯性太大,”周边”太多, 整个生信软件生态没有一致性更新,所以很多分析只能用hg19做。
Cell Ranger ATAC (v 1.1.0) 能做很多工作,
- Read过滤和比对(Read filtering and alignment)
- Barcode记数(Barcode counting)
- 鉴定转座酶切割位点(Identification of transposase cut sites)
- 发现可及染色质峰(Detection of accessible chromatin peaks)
- 定义细胞(Cell calling)
- peak和TF的count矩阵(Count matrix generation for peaks and transcription factors)
- 降维(Dimensionality reduction)
- 细胞聚类(Cell clustering)
- 聚类间差异可及性分析(Cluster differential accessibility)
原文用了很长一段描述(见Data processing using Cell Ranger ATAC software)来说明了Cell Ranger ATAC的具体过程。当然我们不在乎过程,我们只在乎结果。而结果就是下面这些:
- singlecell.csv: 每个细胞的信息,如是否在TSS
- possorted_bam.bam,possorted_bam.bam.bai: 处理后的BAM与其索引
- raw_peak_bc_matrix.h5: 原始的peak-cell矩阵, h5格式存放
- raw_peak_bc_matrix: 原始的peak-cell矩阵
- analysis: 各种分析结果,如聚类,富集,降维等
- filtered_peak_bc_matrix.h5: 过滤后的peak-cell矩阵, h5格式存放
- filtered_peak_bc_matrix: 过滤后的peak-cell矩阵
- fragments.tsv.gz, fragments.tsv.gz.tbi: 每个barcode的序列和它对应的基因组位置和数目
- filtered_tf_bc_matrix.h5: 过滤后的TF-cell矩阵, h5格式存放
- filtered_tf_bc_matrix: 过滤后的TF-cell矩阵
- cloupe.cloupe: Loupe Cell Browser的输入文件
- summary.csv, summary.json: 数据的统计结果, 以csv和json格式存放
- web_summary.html: 网页总结信息
- peaks.bed: 所有的peak汇总
- peak_annotation.csv: peak的注释结果
虽然结果很多,但是文章只用到了framgments.tsv.gz,就如同我们单细胞转录组分析也只要表达量矩阵。
scATAC-seq 数据分析
之后文章的方法部分就开始描述scATAC-seq的分析内容,一共有19项,描述多达3.5页。如果让作者在附录尽情发挥的话,那么每一项都会有1到2页内容,所以这将会一项非常持久的更新计划。
- Filtering cells by TSS enrichment and unique fragments
- Generating a counts matrix
- Generating union peak sets with LSI
- Reads-in-peaks-normalized bigwigs and sequencing tracks
- ATAC-seq-centric LSI clustering and visualization
- Inferring copy number amplification
- TF footprinting
- ChromVAR
- Computing gene activity scores using Cicero co-accessibility
- Analysis of autoimmune variants using Cicero co-accessibility and chromVAR.
- HiChIP meta-virtual 4C (metav4C) analysis for Cicero co-accessibility links
- Overlap of Cicero co-accessibility links with GTEx eQTLs
- Constructing ATAC-seq pseudo-bulk replicates of maximal variance
- Constructing gene score pseudo-bulk replicates of maximal variance
- Identification of cluster-specific peaks and gene scores through feature binarization
- Pseudotime analysis
- Barnyard mixing analysis
- Analysis of fresh versus frozen PBMCs
- Spike-in analysis
根据TSS富集和唯一片段过滤细胞
这一步用到的脚本是01_Filter_Cells_v2.R,读取的fragments为四列。前三列是fragment在染色体的位置,第四列是barcode信息,第五列则是该fragments被测了几次。
1 | #chr start end barcode number |
分析者先过滤不到那些fragments少于100的细胞。
之后,按照文章里提到过滤低质量细胞的标准进行过滤
- cut-offs of 1,000 unique nuclear fragments per cell
- a transcription start site (TSS) enrichment score of 8
每个细胞的唯一片段数计算比较简单也很好理解,而TSS enrichment score需要看他们之前在2018年发表在science文章的附录(Corces et. al 2018 science)
To get the fragment length distribution, the width of each
fragment/GRange was plotted. To get the TSS enrichment profile, each TSS from the R package “TxDb.Hsapiens.UCSC.hg38.knownGene” (accessed by transcripts(TxDb)) was extended 2000 bp in each direction and overlapped with the insertions (each end of a fragment) using “findOverlaps”. Next, the distance between the insertions and the strand-corrected TSS was calculated and the number of insertions occurring in each single-base bin was summed. To normalize this value to the local background, the accessibility at each position +/- 2000 bp from the TSS was normalized to the mean of the accessibility at positions +/-1900-2000 bp from the TSS. The final TSS enrichment reported was the maximum enrichment value within +/- 50 bp of the TSS after smoothing with a rolling mean every 51 bp.
这依赖于一个核心函数,insertionProfileSingles, 作用是分析每个细胞的插入谱
最后分析者为了避免潜在的doublets,还过滤了含有大于45,000的唯一fragmeng的细胞
构建counts矩阵
这部分描述包括后续的各种矩阵产生,包括cell-by-window, cell-by-peak, cell-by-tf。主要涉及02_Get_Peak_Set_hg19_v2.R 里的countInsertions函数。
第一步:获取 fragment GenomicRanges(数据结构如下),也就是01_Filter_Cells_v2.R过滤后的结果
1 | GRanges object with 6 ranges and 2 metadata columns: |
第二步: 然后通过将fragments的起始位置和结束位置进行合并, 就得到了Tn5 insertion GenomicRange
1 | by <- "RG" |
第三步: 用findOverlaps找和insertion重叠的feature(如window, TSS, peak, TF)
1 | overlapDF <- DataFrame(findOverlaps(query, |
第四步: 增加新的一列(id)到重叠输出对象中,这一列的内容是细胞barcode的ID,然后转成sparseMatrix提高存储和计算效率
1 | overlapDF$name <- mcols(inserts)[overlapDF[, 2], by] |
第五步: Tn5插入数在peak的比例 (frip)计算方法为: 稀疏矩阵的列和和每个细胞的fragment数相除
1 | inPeaks <- table(overlapDF$name) |
第六步(可选): counts矩阵用edgeR的cpm(matrix, log = TRUE, prior.count = 3)进行log标准化。这个矩阵用于降低那些低count值元素对变异的影响。该标准化假设不同细胞类型中所有染色质开放区的差异是比较小的。
1 | logMat <- edgeR::cpm(clusterSums, log = TRUE, prior.count = 3) |
利用LSI生成peak合集
方法参考自Cusanovich, D. A. et al (Cell 2018), 代码见02_Get_Peak_Set_hg19_v2.R, 步骤如下
第一步: 利用tile函数,沿着基因组构建2.5kb的window
1 | genome <- BSgenome.Hsapiens.UCSC.hg19 |
第二步: 利用countInsertions基于windows和fragments获取cell-by-window矩阵。
1 | # lapply遍历所有的fragments, 然后合并 |
第三步: 矩阵二项化,并且只要所有细胞中前20,000最开放位点
1 | # seuratLSI |
第四步: 利用TF-IDF(term frequency-inverse document frequency)降维
1 | # 计算单个细胞中每个开放位点的频率 |
TF-IDF的思想: 如果某个词比较少见,但是它在这篇文章中多次出现,那么它很可能就反映了这篇文章的特性,正是我们所需要的关键词 – http://www.ruanyifeng.com/blog/2013/03/tf-idf.html
第五步: 利用R语言中的irlba对标准化TF-IDF进行奇异值分解(SVD) , 保留第2-25个维度(第一个维度通常和cell read depth高度相关)作为Seurat的输入
1 | # SVD降维 |
第三,四,五步被整合到一个函数seuratLSI中, 文章用的是Seurat V2.3
第六步: 并用FindClusters进行SNN图聚类(默认0.8分辨率), 如果最小的细胞类群细胞数不够200,降低分辨率重新聚类, 一个函数addClusters实现。
1 | addClusters <- function(obj, minGroupSize = 50, dims.use = seq_len(50), initialResolution = 0.8){ |
需要注意,文章用的Seurat版本V2.3, 所以如果安装3.0版本, 上面的代码就失效了但是有解决方案,参考如下博客
第七步: 之后用MACS2对每个聚类里 Tn5-corrected singlebase insertions (each end of the Tn5-corrected fragments) 都进行peak calling,参数设置为–shift -75–extsize 150–nomodel–callsummits–nolambda–keep-dup all -q 0.05
1 | # 保存BED文件 |
之后读取peak summits文件,前后延伸250 bp, 最终宽度为501bp, 并基于ENCODE hg19 blacklist(https://www.encodeproject.org/annotations/ENCSR636HFF/)过滤,以及那些延伸后会超出染色体的peak。
1 | # 读取 |
其中extendPeakSet里的函数nonOverlappingGRanges 会对单个样本中相互重叠的peak进行合并处理。步骤如下
- 如果存在有直接重叠的peak,就只保留最显著的peak,过滤不怎么显著的peak
- 迭代上一步,处理下一个重叠的peak,直到这类直接性重叠的peak要么都保留,要么都移除。这一步用于处理每个聚类的peak集,保留前200,000延伸的summit, 得到了每个cluster的’cluster-specific’ peak集
- 接着对每个样本的MACS2 peak score进行标准化,即
−log10(Q value), 然后通过将每个个体的score转成中位数trunc(rank(v))/length(v)’, 其中v表示MACS2 peak scores 向量, 等到”score quantitle”。标准化之后peak就能够在聚类之间进行相互比较,能够产生每个数据集的peak合集 - 合并所有的cluster的peak,使用之前的迭代方法处理重叠,得到了最显著的peak,抛弃了不怎么显著的peak
- 接着将那些和基因组gap区(也就是序列是N)重叠peak移掉,以及Y染色体上的peak
第八步: 构建cell-by-peak矩阵. 用到的是countInsertions函数,只不过第一个参数改为了unionPeaks.
1 | #Create Counts list |
做一些总结性分析
1 | #CountsMatrix |
Reads-in-peaks-normalized bigwigs and sequencing tracks.
这部分内容在GitHub上没有代码,所以我根据文章描述编写代码实现
…
ATAC-seq-centric LSI 聚类和可视化
以Cusanovich et. al (Nature 2018) 提出的策略计算TF-IDF转换, 然后irlba进行奇异值分解(SVD), 接着用前50维作为Seurat的输入,实现SNN图聚类,然后用FindCluster寻找聚类。 你会发现这个内容和上一节类似,只不过上一节是基于window, 这一节是基于peak。
1 | # TF-IDF, SVD |
分析者发现这里存在能被检测到的批次效应,它会混淆后续的分析。因此为了减缓影响,他们从二项化后到可及矩阵中计算每个聚类的和,然后对edgeR的cpm(matrix, log = TRUE, prior.count = 3)结果进行log标准化
1 | mat <- assay(se) |
然后用rowVars挑选前25,000个波动peak,这是基于上一步标准化的矩阵
1 | varPeaks <- head(order(matrixStats::rowVars(logMat), decreasing = TRUE), nTop) |
为什么不用原来的稀疏二项矩阵,而用log标准化的CPM矩阵,原因有亮点
- 能够降低不同聚类间因细胞数不同导致的误差
- count转换到标准化空间后的均值-变异关系会减弱
之后重新用25,000波动peak提取稀疏的二项化矩阵,然后重新做TF-IDF, SVD, SNN graph聚类等运算。
1 | #Re-run Seurat LSI |
用Seurat::RunUMAP函数进行可视化展示
1 | message("Running UMAP") |
对于亚群分析(hematopoiesis: CD34 + bone marrow 和 DCs; tumor: T cells), 分析者重新计算了聚类的和以及log标准化的CPM值,分别从CD34+ cells 和 T cells中鉴定出前10,00和5,00个波动peak。这些波动peak用于提取稀疏二项化可及矩阵,然后做TF-IDF转换。对TF-IDF转换结果进行奇异值分解(SVD),得到更低的维度, 即用前25个维度表征原来的数据集。分贝用1-25和2-25个维度作为Seurat的输入,进行SNN graph clustering(默认resolution=0.8). 最后的结果用Seurat::RunUMAP函数进行可视化展示
细胞类群定义
以human hematopoiesis为例,介绍作者定义细胞类群的三种方法。
(1) 顺式作用元件(cis-lements, 即ATAC-seq peaks)的染色质可及性
(2) 利用和单个基因启动子相关的几个增强子的可及性计算基因活跃得分(GA)
(3) TF活跃度,通过计算每个细胞中全基因组范围内的TF结合位点可及性得出
这三种方法最大的特点就是不需要你再去测一个单细胞RNA-seq或者要一个已有的Bulk ATAC-seq谱,仅仅用当前的单细胞ATAC-seq数据即可。