如何用软件模拟NGS数据
为了评价一个工具的性能,通常我们都需要先模拟一批数据。这样相当于有了参考答案,才能检查工具的实际表现情况。因此对于我们而言,面对一个新的功能,可以先用模拟的数据测试下不同工具的优缺点。有如下几个工具值得推荐一下:
- ‘wgsim/dwgsim’: 从全基因组中获取测序reads
- ‘msbar’: EMBOSS其中一个工具,能够从单个序列中模拟随机突变
- ‘biosed’: EMBOSS的一个工具,可以按照我们给定突变位点模拟
- ‘ReadSim’: 专门用于模拟PacBio/Nanopore这类仪器产生的long read
- ‘Art’: 目前最复杂的模拟工具,能够模拟测序仪测序引入的错误位点
- ‘Metasim’: 用于模拟宏基因组得到的reads
- ‘Polyester’: 用于模拟RNA-seq
值得注意的是,这些工具模拟效果是有限,比如建库操作中超声破碎会出现的误差就很难模拟。但是最好的用途就是看看不同生物学事件在数据的情况,比如说发生了“大规模倒置”的基因组得到的数据比对到参考基因组上会是什么情况。
使用dwgsim进行模拟
wgism和dwgsim能够根据参考基因组模拟出测序reads,主要是二倍体基因组的SNPs和插入缺失(INDEL)多态位点。wgism容易安装,但是参考答案是以简单的文本格式保存,不容易可视化。dwgsim受wgism启发,虽然安装稍微麻烦了点,但是参考答案是以VCF格式保存,很方便可视化。
1 | # 请先安装好ncurse |
简单地模拟一批数据
1 | # efetch 需要用到conda安装启动 |
会得到如下数据
1 | |-- data.bfast.fastq.gz # 用于bfast |
随后将这批数据用BWA比对,以bcftools检测变异和参考答案比较一下。
1 | # conda install bwa samtools bcftools |
利用使用IGV可视化,检查分析结果和真集的一致性
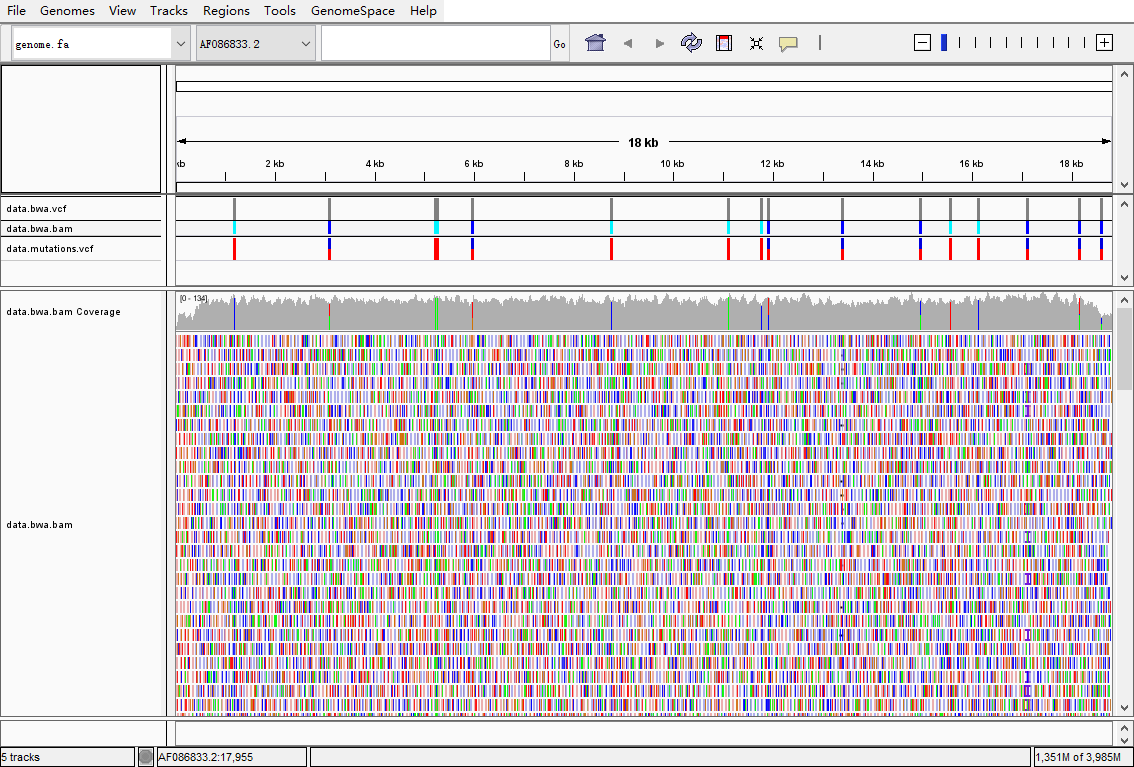
说明samtools+bcftools找变异这个组合其实还是靠谱的,至少在动植物领域研究里应该够用。