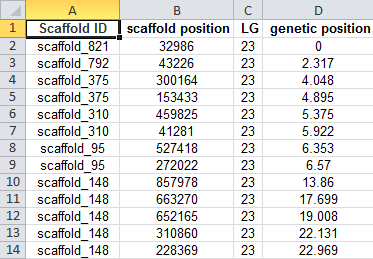
为了避免「一学就会,一写就废」,因此我给自己设置了一个练习题,就是读取一个只有两列的CSV文件,用第一列当做key,第一列当做value,保存到HashMap(哈希表)中。
用Python完成这个任务非常的简单,代码如下
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 import csvimport syscsv_file = sys.argv[1 ] hash_map = {} with open (csv_file, "r" ) as f: for line in csv.reader(f): key = line[0 ] value = line[1 ] if key in hash_map: hash_map[key].append(value) else : hash_map[key] = [ value ]
根据Python的代码,我查找了rust中csv和HashMap的相关文档并学习,但是我对Rust本身的语言特性并不是特别熟悉,导致花了一天才处理完报错并理解背后的逻辑。
Rust的变量默认不可变 和我之前学的C/C++, Python, R等编程语言不同,Rust的变量默认是不可改变的,也就是下面的代码是错误
1 2 let hm = HashMap::new ();hm.insert ("key" ,"value" );
你必须显式用关键词 mut声明,Rust才允许你对一个变量进行改变。
1 2 3 let mut hm = HashMap::new ();hm.insert ("key" ,"value" );
一开始觉得很不习惯,但是后来想想也挺合理的。如果一个变量,你没有改变它的想法,但是你编写的某些代码却能改动它,就可能引起bug。同时,如果一个值,你认为它需要改变,但是实际上它没有变动,那就意味着你应该去掉这个mut声明。
Rust要求你时刻考虑错误处理。 Rust通过类型系统来处理异常情况,因此许多函数并不会直接返回结果,而是返回Option或Result。这就导致我们从函数里得到的值无法直接使用,需要多写一点代码从中提取目标结果。
下面代码中csv_reader的类型是 Result<Reader<File>, Error>, 因此无法使用for循环进行遍历。
1 2 3 4 5 use csv::Reader;let mut csv_reader = Reader::from_path (csv_file);for result in csv_reader.records () { .... }
处理方法有两种,一种是使用?操作符 将错误往后传播处理
1 2 3 for result in csv_reader.records ()? { .... }
另一种则是调用.unwrap()直接退出程序,
1 2 3 for result in csv_reader.records ().unwrap () { .... }
同样的,遍历得到result也不能直接使用,需要通过模式匹配,从中提取出目标结果
1 2 3 4 if let Ok (record) = result{ println! ("{}" , record); ... }
Rust要求你注意值和借用的生命周期 在Rust中,每个值都有其生命周期,对值的引用(借用)不能超过(outlive)值的生存期。
在下面代码中,我的目标是遍历每一行,然后将x进行split构建成一个vector,然后提取k,v,进行插入
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 fn main () -> io::Result <()>{ let args : Vec <String > = env::args ().collect (); let csv_file = &args[1 ]; let mut hm = HashMap::new (); for line in io::BufReader::new (fs::File::open (csv_file)?).lines (){ if let Ok (x) = line { let s : Vec <&str > = x.split (',' ).collect (); let k = s[0 ]; let v = s[1 ]; hm.insert (k, v); } } Ok (()) }
但是代码会报错,出现如下提示
1 2 3 4 5 6 7 error[E0597]: `x` does not live long enough let s : Vec <&str > = x.split (',' ).collect (); ^ borrowed value does not live long enough hm.insert (s[0 ],s[1 ]); -- borrow later used here } - `x` dropped here while still borrowed
从我的角度看,虽然x是被借用的数据,但是通过split和collect得到的s,里面记录的 &str指向的字符串应该是 新分配的,跟x没有关系才对,所以我觉得就算是报错,也应该出在s的生命周期上。但实际上,通过对文档的阅读,我才知道vector里存放的值实际是x的切片,每一次循环结束后,x的生命周期就结束了,切片数据也同样不复存在。
An iterator over substrings of this string slice , separated by characters matched by a pattern.
同时由于&str只是字符串的借用,因此无法转移所有权, 需要将&str转成String才行。
1 2 3 4 5 6 7 8 9 10 11 12 let k = s[0 ].to_string ();let v = s[1 ].to_string ();
如果用的csv库进行解析,也同样需要考虑这个问题。下面代码会因为record的生命周期出错,也需要将其转成String。
1 2 3 4 5 6 7 8 9 10 11 let mut rdr = ReaderBuilder::new ().from_path (csv_file)?;for result in rdr.records (){ if let Ok (record) = result{ let k = record.get (0 ).unwrap (); let v = record.get (1 ).unwrap (); let vec = hm.entry (k).or_insert (Vec ::new ()); vec.push (v); } }
除开上面两个主要问题外,还有一些小问题,也简单记录一下
Rust并不总能推导出变量的类型,比如说由于有很多集合类型都实现了collect方法,就导致Rust不知道你调用collect到底要什么
Rust的vector添加单个元素是push, append是合并另外一个vector
HashMap的get函数只能返回key所对应的value的引用,改引用无法修改,get_mut返回才是可变引用
HashMap除了使用get_mut函数获取可变引用进行修改外,还可以使用entry函数。
最终代码如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 use std::error::Error;use std::env;use std::collections::HashMap;use csv::Reader;fn main () -> Result <(), Box <dyn Error>>{ let args : Vec <String > = env::args ().collect (); let csv_file = &args[1 ]; let mut hm = HashMap::new (); let mut rdr = Reader::from_path (csv_file)?; for result in rdr.records (){ if let Ok (record) = result{ let k = record.get (0 ).unwrap (); let v = record.get (1 ).unwrap (); let vec = hm.entry (k).or_insert (Vec ::new ()); vec.push (v); } } for (key, value) in &hm{ println! ("{}: {:?}" , key, value); } Ok (()) }